Next Generation xcms Result Object
Source: R/AllGenerics.R, R/MsExperiment.R, R/XcmsExperiment-plotting.R, and 2 more
XcmsExperiment.RdThe XcmsExperiment is a data container for xcms preprocessing
results (i.e. results from chromatographic peak detection, alignment and
correspondence analysis). It is the preferred and default result object
since version 4 of xcms.
It provides the same functionality than the XCMSnExp object, but uses the more advanced and modern MS infrastructure provided by the MsExperiment and Spectra Bioconductor packages. This enables a much higher flexibility of data representation and storage and ensures future expandability.
Documentation of the various functions for XcmsExperiment objects are
grouped by topic and provided in the sections below.
The default xcms data analysis workflow is to perform:
chromatographic peak detection using
findChromPeaks()optionally refine identified chromatographic peaks using
refineChromPeaks()(this is highly suggested for centWave-based chromatographic peak detection)retention time alignment (retention time adjustment) using
adjustRtime(). Depending on the method used, this may require to run a correspondence analysis firstcorrespondence analysis to group chromatographic peaks across samples to define the LC-MS features using the
groupChromPeaks()functiongap-filling to rescue signal in samples in which no chromatographic peak was identified and hence a missing value would be reported. This can be performed using the
fillChromPeaks()function.
For very large LC-MS experiments (either with a very large number of samples
or very large data files, or both), the XcmsExperimentHdf5() object can
be used instead. See the respective help page for more information.
Usage
filterFeatureDefinitions(object, ...)
# S4 method for class 'MsExperiment'
filterRt(object, rt = numeric(), ...)
# S4 method for class 'MsExperiment'
filterMzRange(object, mz = numeric(), msLevel. = uniqueMsLevels(object))
# S4 method for class 'MsExperiment'
filterMz(object, mz = numeric(), msLevel. = uniqueMsLevels(object))
# S4 method for class 'MsExperiment'
filterMsLevel(object, msLevel. = uniqueMsLevels(object))
# S4 method for class 'MsExperiment'
uniqueMsLevels(object)
# S4 method for class 'MsExperiment'
filterFile(object, file = integer(), ...)
# S4 method for class 'MsExperiment'
rtime(object)
# S4 method for class 'MsExperiment'
fromFile(object)
# S4 method for class 'MsExperiment'
fileNames(object)
# S4 method for class 'MsExperiment'
polarity(object)
# S4 method for class 'MsExperiment'
filterIsolationWindow(object, mz = numeric())
# S4 method for class 'MsExperiment'
chromatogram(
object,
rt = matrix(nrow = 0, ncol = 2),
mz = matrix(nrow = 0, ncol = 2),
aggregationFun = "sum",
msLevel = 1L,
isolationWindowTargetMz = NULL,
chunkSize = 2L,
return.type = "MChromatograms",
BPPARAM = bpparam()
)
# S4 method for class 'MsExperiment,missing'
plot(x, y, msLevel = 1L, peakCol = "#ff000060", ...)
# S3 method for class 'XcmsExperiment'
c(...)
# S4 method for class 'XcmsExperiment,ANY,ANY,ANY'
x[i, j, ..., drop = TRUE]
# S4 method for class 'XcmsExperiment'
filterIsolationWindow(object, mz = numeric())
# S4 method for class 'XcmsExperiment'
filterRt(object, rt, msLevel.)
# S4 method for class 'XcmsExperiment'
filterMzRange(object, mz = numeric(), msLevel. = uniqueMsLevels(object))
# S4 method for class 'XcmsExperiment'
filterMsLevel(object, msLevel. = uniqueMsLevels(object))
# S4 method for class 'XcmsExperiment'
hasChromPeaks(object, msLevel = integer())
# S4 method for class 'XcmsExperiment'
dropChromPeaks(object, keepAdjustedRtime = FALSE)
# S4 method for class 'XcmsExperiment'
chromPeaks(object) <- value
# S4 method for class 'XcmsExperiment'
chromPeaks(
object,
rt = numeric(),
mz = numeric(),
ppm = 0,
msLevel = integer(),
type = c("any", "within", "apex_within"),
isFilledColumn = FALSE,
columns = character()
)
# S4 method for class 'XcmsExperiment'
chromPeakData(object) <- value
# S4 method for class 'XcmsExperiment'
chromPeakData(
object,
msLevel = integer(),
columns = character(),
return.type = c("DataFrame", "data.frame")
)
# S4 method for class 'XcmsExperiment'
filterChromPeaks(
object,
keep = rep(TRUE, nrow(.chromPeaks(object))),
method = "keep",
...
)
# S4 method for class 'XcmsExperiment'
dropAdjustedRtime(object)
# S4 method for class 'MsExperiment'
hasAdjustedRtime(object)
# S4 method for class 'XcmsExperiment'
rtime(object, adjusted = hasAdjustedRtime(object))
# S4 method for class 'XcmsExperiment'
adjustedRtime(object)
# S4 method for class 'XcmsExperiment'
hasFeatures(object, msLevel = integer())
# S4 method for class 'XcmsResult'
featureArea(
object,
mzmin = min,
mzmax = max,
rtmin = min,
rtmax = max,
features = character(),
minMzWidthPpm = 0
)
# S4 method for class 'XcmsExperiment'
featureDefinitions(object) <- value
# S4 method for class 'XcmsExperiment'
featureDefinitions(
object,
mz = numeric(),
rt = numeric(),
ppm = 0,
type = c("any", "within", "apex_within"),
msLevel = integer()
)
# S4 method for class 'XcmsExperiment'
dropFeatureDefinitions(object, keepAdjustedRtime = FALSE)
# S4 method for class 'XcmsExperiment'
filterFeatureDefinitions(object, features = integer())
# S4 method for class 'XcmsExperiment'
hasFilledChromPeaks(object)
# S4 method for class 'XcmsExperiment'
dropFilledChromPeaks(object)
# S4 method for class 'XcmsExperiment'
quantify(object, ...)
# S4 method for class 'XcmsExperiment'
featureValues(
object,
method = c("medret", "maxint", "sum"),
value = "into",
intensity = "into",
filled = TRUE,
missing = NA_real_,
msLevel = integer()
)
# S4 method for class 'XcmsExperiment'
chromatogram(
object,
rt = matrix(nrow = 0, ncol = 2),
mz = matrix(nrow = 0, ncol = 2),
aggregationFun = "sum",
msLevel = 1L,
chunkSize = 2L,
isolationWindowTargetMz = NULL,
return.type = c("XChromatograms", "MChromatograms"),
include = character(),
chromPeaks = c("apex_within", "any", "none"),
BPPARAM = bpparam()
)
# S4 method for class 'XcmsExperiment'
processHistory(object, type)
# S4 method for class 'XcmsExperiment'
filterFile(
object,
file,
keepAdjustedRtime = hasAdjustedRtime(object),
keepFeatures = FALSE,
...
)Arguments
- object
An
XcmsExperimentobject.- ...
Additional optional parameters. For
quantify(): any parameter for thefeatureValuescall used to extract the feature value matrix.- rt
For
chromPeaks()andfeatureDefinitions():numeric(2)defining the retention time range for which chromatographic peaks or features should be returned. The full range is used by default. Forchromatogram(): two column numericalmatrixwith each row representing the lower and upper retention time window(s) for the chromatograms. If not provided the full retention time range is used.- mz
For
chromPeaks()andfeatureDefinitions():numeric(2)optionally defining the m/z range for which chromatographic peaks or feature definitions should be returned. The full m/z range is used by default. Forchromatogram(): two-column numericalmatrixwith each row representing m/z range that should be aggregated into a chromatogram. If not provided the full m/z range of the data will be used (and hence a total ion chromatogram will be returned ifaggregationFun = "sum"is used). ForfilterIsolationWindow():numeric(1)defining the m/z that should be contained within the spectra's isolation window.- msLevel.
For
filterRt(): ignored.filterRt()will always filter by retention times on all MS levels regardless of this parameter. Forchromatogram():integerwith the MS level from which the chromatogram(s) should be extracted. Has to be either of length 1 or length equal to the numer of rows of the parametersmzandrtdefining the m/z and rt regions from which the chromatograms should be created. Defaults tomsLevel = 1L. forfilterMsLevel():integerdefining the MS level(s) to which the data should be subset.- file
For
filterFile():integerwith the indices of the samples (files) to which the data should be subsetted.- aggregationFun
For
chromatogram():character(1)defining the function that should be used to aggregate intensities for retention time (i.e. each spectrum) along the specified m/z range (parametermz). Defaults toaggregationFun = "sum"and hence all intensities will be summed up. Alternatively, useaggregationFun = "max"to use the maximal intensity per m/z range to create a base peak chromatogram (BPC).- msLevel
integerdefining the MS level (or multiple MS level if the function supports it).- isolationWindowTargetMz
For
chromatogram():numeric(of length equal to the number of rows ofrtandmz) with the isolation window target m/z of the MS2 spectra from which the chromatgrom should be generated. For MS1 data (msLevel = 1L, the default), this parameter is ignored. See examples onchromatogram()below for further information.- chunkSize
For
chromatogram():integer(1)defining the number of files from which the data should be loaded at a time into memory. Defaults tochunkSize = 2L.- return.type
For
chromPeakData():character(1)defining the class of the returned object. Can be either"DataFrame"(the default) or"data.frame". Forchromatogram():character(1)defining the type of the returned object. Currently onlyreturn.type = "MChromatograms"is supported.- BPPARAM
For
chromatogram(): parallel processing setup. Defaults toBPPARAM = bpparam(). SeeBiocParallel::bpparam()for more information.- x
An
XcmsExperimentobject.- y
For
plot(): should not be defined as it is not supported.- peakCol
For
plot(): defines the border color of the rectangles indicating the identified chromatographic peaks. Only a single color is supported. Defaults to `peakCol = "#ff000060".- i
For
[:integerorlogicaldefining the samples/files to subset.- j
For
[: not supported.- drop
For
[: ignored.- keepAdjustedRtime
logical(1): whether adjusted retention times (if present) should be retained.- value
For
featureValues():character(1)defining which value should be reported for each feature in each sample. Can be any column of thechromPeaks()matrix or"index"if simply the index of the assigned peak should be returned. Defaults tovalue = "into"thus the integrated peak area is reported.- ppm
For
chromPeaks()andfeatureDefinitions(): optionalnumeric(1)specifying the ppm by which the m/z range (defined bymzshould be extended. For a value ofppm = 10, all peaks withinmz[1] - ppm / 1e6andmz[2] + ppm / 1e6are returned.- type
For
chromPeaks()andfeatureDefinitions()and only if eithermzandrtare defined too:character(1): defining which peaks (or features) should be returned. Fortype = "any": returns all chromatographic peaks or features also only partially overlapping any of the provided ranges. Fortype = "within": returns only peaks or features completely within the region defined bymzand/orrt. Fortype = "apex_within": returns peaks or features for which the m/z and retention time of the peak's apex is within the region defined bymzand/orrt. ForprocessHistory(): restrict returned processing steps to specific types. UseprocessHistoryTypes()to list all supported values.- isFilledColumn
For
chromPeaks():logical(1)whether a column"is_filled"should be included in the returnedmatrixwith the information whether a peak was detected or only filled-in. Note that this information is also provided in thechromPeakDatadata frame.- columns
For
chromPeaks()andchromPeakData(): optionalcharacterto specify the names of the columns that should be returned. By default (withcolumns = character()all columns are returned.- keep
For
filterChromPeaks():logical,integerorcharacterspecifying which chromatographic peaks to keep. Iflogicalthe length ofkeepneeds to match the number of rows ofchromPeaks(). Alternatively,keepallows to specify theindex(row) of peaks to keep or their ID (i.e. row name inchromPeaks()).- method
For
featureValues():character(1)specifying the method to resolve multi-peak mappings within the same sample (correspondence analysis can assign more than one chromatographic peak within a sample to the same feature, e.g. if they are close in retention time). Options:method = "medret": report the value for the chromatographic peak closest to the feature's median retention time.method = "maxint": report the value for the chromatographic peak with the largest signal (parameterintensityallows to select the column inchromPeaksthat should be used for signal).method = "sum": sum the value for all chromatographic peaks in a sample assigned to the same feature. The default ismethod = "medret". ForfilterChromPeaks(): currently onlymethod = "keep"is supported.- adjusted
For
rtime,XcmsExperiment: whether adjusted or raw retention times should be returned. The default is to return adjusted retention times, if available.- mzmin
For
featureArea(): function to calculate the"mzmin"of a feature based on the"mzmin"values of the individual chromatographic peaks assigned to that feature. Defaults tomzmin = min.- mzmax
For
featureArea(): function to calculate the"mzmax"of a feature based on the"mzmax"values of the individual chromatographic peaks assigned to that feature. Defaults tomzmax = max.- rtmin
For
featureArea(): function to calculate the"rtmin"of a feature based on the"rtmin"values of the individual chromatographic peaks assigned to that feature. Defaults tortmin = min.- rtmax
For
featureArea(): function to calculate the"rtmax"of a feature based on the"rtmax"values of the individual chromatographic peaks assigned to that feature. Defaults tortmax = max.- features
For
filterFeatureDefinitions()andfeatureArea():logical,integerorcharacterdefining the features to keep or from which to extract the feature area, respectively. See function description for more information.- minMzWidthPpm
For
featureArea():numeric(1)defining the minimal guaranteed m/z width (expressed in ppm of the feature's m/z) of the reported feature areas. Defaults tominMzWidthPpm = 0.0. See documentation of thefeatureArea()for more information.- intensity
For
featureValues():character(1)specifying the name of the column in thechromPeaks(objects)matrix containing the intensity value of the peak that should be used for the conflict resolution ifmethod = "maxint".- filled
For
featureValues():logical(1)specifying whether values for filled-in peaks should be reported. Forfilled = TRUE(the default) filled peak values are returned, otherwiseNAis reported for the respective features in the samples in which no peak was detected.- missing
For
featureValues(): default value for missing values. Allows to define the value that should be reported for a missing peak intensity. Defaults tomissing = NA_real_.- include
For
chromatogram(): deprecated; use parameterchromPeaksinstead.- chromPeaks
For
chromatogram():character(1)defining which chromatographic peaks should be returned. Can be eitherchromPeaks = "apex_within"(default) to return all chromatographic peaks with the m/z and RT of their apex within the m/z and retention time window,chromPeaks = "any"for all chromatographic peaks that are overlapping with the m/z - retention time window orchromPeaks = "none"to not include any chromatographic peaks. See also parametertypebelow for additional information.- keepFeatures
for most subsetting functions (
[,filterFile()):logical(1): wheter eventually present feature definitions should be retained in the returned (filtered) object.
Subset, filter and combine
[: subset anXcmsExperimentby sample (parameteri). Subsetting will by default drop correspondence results (as subsetting by samples will obviously affect the feature definition) while alignment results (adjusted retention times) and identified chromatographic peaks (for the selected samples) will be retained. Which preprocessing results should be kept or dropped can also be configured with optional parameterskeepChromPeaks(by defaultTRUE),keepAdjustedRtime(by defaultTRUE) andkeepFeatures(by defaultFALSE).c(): multipleXcmsExperimentobjects can be combined into one using thec()function. This requires however that all theXcmsExperiments'Spectraobjects use the same type ofMsBackendand that their processing queues are empty. Also, only combining of peak detection results is supported. Any eventually present alignment or correspondence results will be dropped before combining theXcmsExperimentobjects. Finally, at present, only the MS data of the individualXcmsExperimentobjects is combined and any data eventually present in the@qdata,@otherDataand@experimentFilesslots is ignored. The function returns aXcmsExperimentobjects with the combined MS data (Spectraobjects) and chromatographic peak detection results.filterChromPeaks(): filter chromatographic peaks of anXcmsExperimentkeeping only those specified with parameterkeep. Returns theXcmsExperimentwith the filtered data. Chromatographic peaks to retain can be specified either by providing their index in thechromPeaks()matrix, their ID (rowname inchromPeaks()) or with alogicalvector with the same length than number of rows ofchromPeaks(). Assignment of chromatographic peaks are updated to eventually present feature definitions after filtering.filterFeatureDefinitions(): filter feature definitions of anXcmsExperimentkeeping only those defined with parameterfeatures, which can be alogicalof length equal to the number of features, anintegerwith the index of the features infeatureDefinitions(object)to keep or acharacterwith the feature IDs (i.e. row names infeatureDefinitions(object)).filterFile(): filter anXcmsExperiment(orMsExperiment) by file (sample). The index of the samples to which the data should be subsetted can be specified with parameterfile. The sole purpose of this function is to provide backward compatibility with theMSnbasepackage. Wherever possible, the[function should be used instead for any sample-based subsetting. ParameterskeepChromPeaks,keepAdjustedRtimeandkeepChromPeakscan be passed using.... Note also that in contrast to[,filterFile()does not support subsetting in arbitrary order.filterIsolationWindow(): filter the spectra within anMsExperimentorXcmsExperimentobject keeping only those with an isolation window containing the specified m/z (i.e., keeping spectra with an"isolationWindowLowerMz"smaller than the user-providedmzand an"isolationWindowUpperMz"larger thanmz). For anXcmsExperimentalso all chromatographic peaks (and subsequently also features) are removed for which the range of their"isolationWindowLowerMz"and"isolationWindowUpperMz"(columns inchromPeakData()) do not contain the user providedmz.filterMsLevel(): filter the data of theXcmsExperimentorMsExperimentto keep only data of the MS level(s) specified with parametermsLevel..filterMz(),filterMzRange(): filter the spectra within anXcmsExperimentorMsExperimentto the specified m/z range (parametermz). ForXcmsExperimentalso identified chromatographic peaks and features are filtered keeping only those that are within the specified m/z range (i.e. for which the m/z of the peak apex is within the m/z range). ParametermsLevels.allows to restrict the filtering to only specified MS levels. By default data from all MS levels are filtered.filterRt(): filter anXcmsExperimentkeeping only data within the specified retention time range (parameterrt). This function will keep all preprocessing results present within the retention time range: all identified chromatographic peaks with the retention time of the apex position within the retention time rangertare retained along, if present, with the associated features. ParametermsLevel.is currently ignored, i.e. filtering will always performed on all MS levels of the object.
Functionality related to chromatographic peaks
chromatogram(): extract chromatographic data from a data set. Parametersmzandrtallow to define specific m/z - retention time regions to extract the data from (to e.g. for extracted ion chromatograms EICs). Both parameters are expected to be numerical two-column matrices with the first column defining the lower and the second the upper margin. Each row can define a separate m/z - retention time region. Currently the function returns aMSnbase::MChromatograms()object forobjectbeing aMsExperimentor, forobjectbeing anXcmsExperiment, either aMChromatogramsorXChromatograms()depending on parameterreturn.type(can be either"MChromatograms"or"XChromatograms"). For the latter also chromatographic peaks detected within the provided m/z and retention times are returned. ParameterchromPeaksallows to specify which chromatographic peaks should be reported. See documentation on thechromPeaksparameter for more information. If theXcmsExperimentcontains correspondence results, also the associated feature definitions will be included in the returnedXChromatograms. By default the function returns chromatograms from MS1 data, but by setting parametermsLevel = 2Lit is possible to e.g. extract also MS2 chromatograms. By default, with parameterisolationWindowTargetMz = NULLorisolationWindowTargetMz = NA_real_, data from all MS2 spectra will be considered in the chromatogram extraction. If MS2 data was generated within different m/z isolation windows (such as e.g. with Scies SWATH data), the parameterisolationWindowTargetMzshould be used to ensure signal is only extracted from the respective isolation window. TheisolationWindowTargetMz()function on theSpectraobject can be used to inspect/list available isolation windows of a data set. See also the xcms LC-MS/MS vignette for examples and details.chromPeaks(): returns anumericmatrix with the identified chromatographic peaks. Each row represents a chromatographic peak identified in one sample (file). The number of columns depends on the peak detection algorithm (seefindChromPeaks()) but most methods return the following columns:"mz"(intensity-weighted mean of the m/z values of all mass peaks included in the chromatographic peak),"mzmin"( smallest m/z value of any mass peak in the chromatographic peak),"mzmax"(largest m/z value of any mass peak in the chromatographic peak),"rt"(retention time of the peak apex),"rtmin"(retention time of the first scan/mass peak of the chromatographic peak),"rtmax"(retention time of the last scan/mass peak of the chromatographic peak),"into"(integrated intensity of the chromatographic peak),"maxo"(maximal intensity of any mass peak of the chromatographic peak),"sample"(index of the sample inobjectin which the peak was identified). Parametersrt,mz,ppm,msLevelandtypeallow to extract subsets of identified chromatographic peaks from theobject. Parametercolumnsallows to optionally define which columns to extract. See parameter description below for details.chromPeakData(): returns aDataFramewith potential additional annotations for the identified chromatographic peaks. Each row in thisDataFramecorresponds to a row (same index and row name) in thechromPeaks()matrix. The default annotations are"ms_level"(the MS level in which the peak was identified) and"is_filled"(whether the chromatographic peak was detected (byfindChromPeaks()) or filled-in (byfillChromPeaks()). Parametercolumnscan be used to restrict the returned data frame to selected columns. Parameterreturn.typecan be used to specify the type of returned objects, either aDataFrame(the default,return.type = "DataFrame") or adata.frame(return.type = "data.frame").chromPeakSpectra(): extract MS spectra for identified chromatographic peaks. This can be either all (full scan) MS1 spectra with retention times between the retention time range of a chromatographic peak, all MS2 spectra (if present) with a retention time within the retention time range of a (MS1) chromatographic peak and a precursor m/z within the m/z range of the chromatographic peak or single, selected spectra depending on their total signal or highest signal. ParametermsLevelallows to define from which MS level spectra should be extracted, parametermethodallows to define if all or selected spectra should be returned. SeechromPeakSpectra()for details.dropChromPeaks(): removes (all) chromatographic peak detection results fromobject. This will also remove any correspondence results (i.e. features) and eventually present adjusted retention times from the object if the alignment was performed after the peak detection. Alignment results (adjusted retention times) can be retained if parameterkeepAdjustedRtimeis set toTRUE.dropFilledChromPeaks(): removes chromatographic peaks added by gap filling withfillChromPeaks().fillChromPeaks(): perform gap filling to integrate signal missing values in samples in which no chromatographic peak was found. This depends on correspondence results, hencegroupChromPeaks()needs to be called first. For details and options seefillChromPeaks().findChromPeaks: perform chromatographic peak detection. SeefindChromPeaks()for details.hasChromPeaks(): whether the object contains peak detection results. ParametermsLevelallows to check whether peak detection results are available for the specified MS level(s).hasFilledChromPeaks(): whether gap-filling results (i.e., filled-in chromatographic peaks) are present.manualChromPeaks(): manually add chromatographic peaks by defining their m/z and retention time ranges. SeemanualChromPeaks()for details and examples.plotChromPeakImage(): show the density of identified chromatographic peaks per file along the retention time. SeeplotChromPeakImage()for details.plotChromPeaks(): indicate identified chromatographic peaks from one sample in the RT-m/z space. SeeplotChromPeaks()for details.plotPrecursorIons(): general visualization of precursor ions of LC-MS/MS data. SeeplotPrecursorIons()for details.refineChromPeaks(): refines identified chromatographic peaks inobject. SeerefineChromPeaks()for details.
Functionality related to alignment
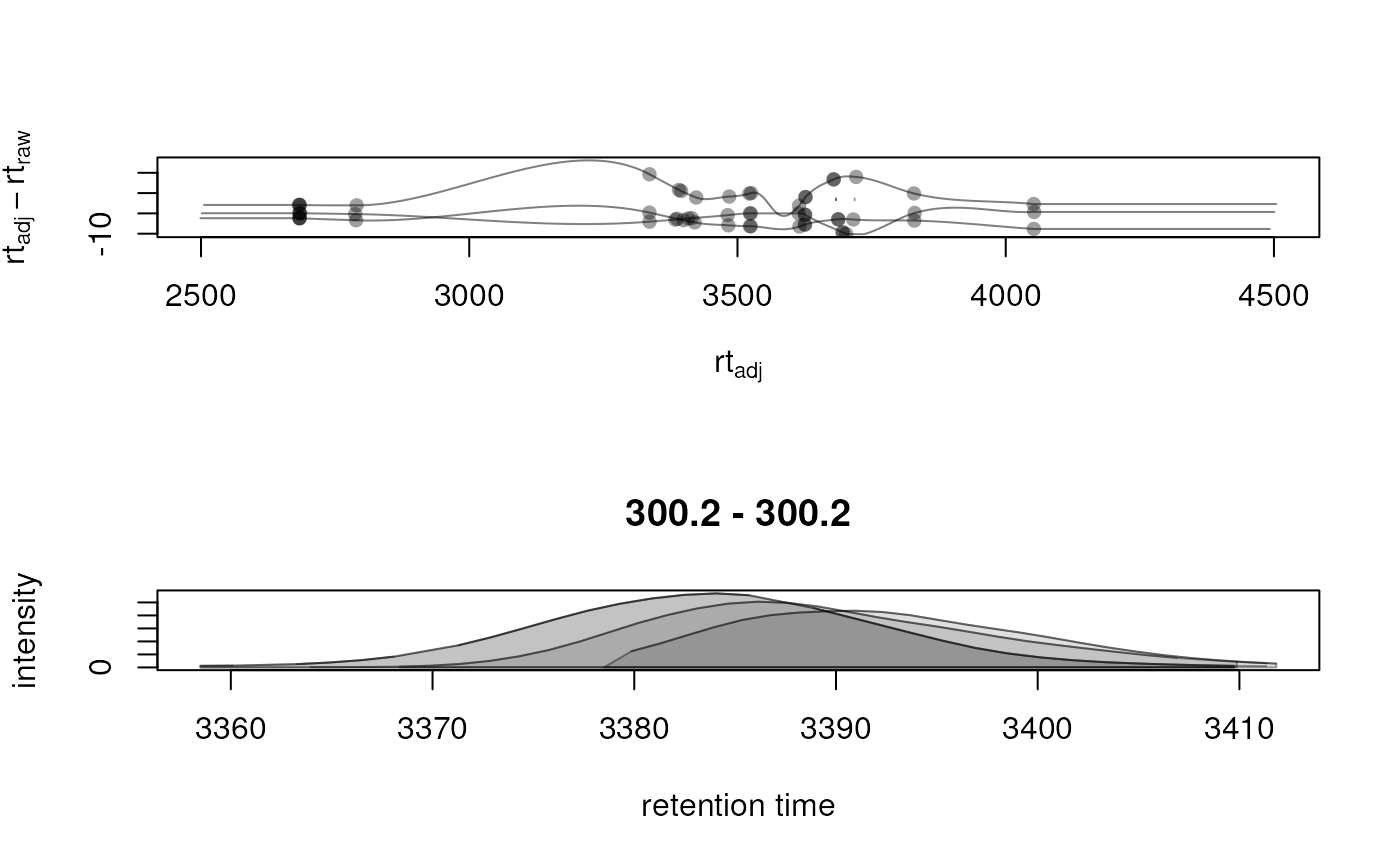
adjustedRtime(): extract adjusted retention times. This is just an alias forrtime(object, adjusted = TRUE).adjustRtime(): performs retention time adjustment (alignment) of the data. SeeadjustRtime()for details.applyAdjustedRtime(): replaces the original (raw) retention times with the adjusted ones. SeeapplyAdjustedRtime()for more information.dropAdjustedRtime(): drops alignment results (adjusted retention time) from the result object. This also reverts the retention times of identified chromatographic peaks if present in the result object. Note that any results from a correspondence analysis (i.e. feature definitions) will be dropped too (if the correspondence analysis was performed after the alignment). This can be overruled withkeepAdjustedRtime = TRUE.hasAdjustedRtime(): whether alignment was performed on the object (i.e., the object contains alignment results).plotAdjustedRtime(): plot the alignment results; seeplotAdjustedRtime()for more information.
Functionality related to correspondence analysis
dropFeatureDefinitions(): removes any correspondence analysis results fromobjectas well as any filled-in chromatographic peaks. By default (with parameterkeepAdjustedRtime = FALSE) also all alignment results will be removed if alignment was performed after the correspondence analysis. This can be overruled withkeepAdjustedRtime = TRUE.featureArea(): returns amatrixwith columns"mzmin","mzmax","rtmin"and"rtmax"with the m/z and retention time range for each feature (row) inobject. By default these represent the minimal m/z and retention times as well as maximal m/z and retention times for all chromatographic peaks assigned to that feature. ParameterminMzWidthPpm(defaultminMzWidthPpm = 0.01) can be used to define a minimal required (total) m/z width expressed in ppm of the features' m/z. With aminMzWidthPpmlarger than 0 the reported"mzmin"is the minimum of the determined minimal m/z for a feature (based on parametermzmin) and the m/z of the feature minusminMzWidthPpm / 2ppm of the feature's m/z value. The reported"mzmax"is calculated in the same way. Parameterfeaturesallows to extract these values for selected features only. Parametersmzmin,mzmax,rtminandrtmaxallow to define the function to calculate the reported"mzmin","mzmax","rtmin"and"rtmax"` values.featureChromatograms(): extract ion chromatograms (EICs) for each feature inobject. SeefeatureChromatograms()for more details.featureDefinitions(): returns adata.framewith feature definitions or an emptydata.frameif no correspondence analysis results are present. ParametersmsLevel,mz,ppmandrtallow to define subsets of feature definitions that should be returned with the parametertypedefining how these parameters should be used to subset the returneddata.frame. See parameter descriptions for details.featureSpectra(): returns aSpectra::Spectra()orListofSpectrawith (MS1 or MS2) spectra associated to each feature's chromatographic peaks. SeefeatureSpectra()for more details and available parameters.featuresSummary(): calculate a simple summary on features. SeefeatureSummary()for details.groupChromPeaks(): performs the correspondence analysis (i.e., grouping of chromatographic peaks into LC-MS features). SeegroupChromPeaks()for details.hasFeatures(): whether correspondence analysis results are presentin inobject. The optional parametermsLevelallows to define the MS level(s) for which it should be determined if feature definitions are available.overlappingFeatures(): identify features that overlapping or close in m/z - rt dimension. SeeoverlappingFeatures()for more information.
Extracting data and results from an XcmsExperiment
Preprocessing results can be extracted using the following functions:
chromPeaks(): extract identified chromatographic peaks. See section on chromatographic peak detection for details.featureDefinitions(): extract the definition of features (chromatographic peaks grouped across samples). See section on correspondence analysis for details.featureValues(): extract amatrixof values for features from each sample (file). Rows are features, columns samples. Which value should be returned can be defined with parametervalue, which can be any column of thechromPeaks()matrix. By default (value = "into") the integrated chromatographic peak intensities are returned. With parametermsLevelit is possible to extract values for features from certain MS levels. During correspondence analysis, more than one chromatographic peak per sample can be assigned to the same feature (e.g. if they are very close in retention time). Parametermethodallows to define the strategy to deal with such cases:method = "medret": report the value from the chromatographic peak with the apex position closest to the feautre's median retention time.method = "maxint": report the value from the chromatographic peak with the largest signal (parameterintensityallows to define the column inchromPeaksthat should be selected; defaults tointensity = "into").method = "sum"`: sum the values for all chromatographic peaks assigned to the feature in the same sample.quantify(): extract the correspondence analysis results as aSummarizedExperiment::SummarizedExperiment(). The feature values are used asassayin the returnedSummarizedExperiment,rowDatacontains thefeatureDefinitions(without column"peakidx") andcolDatathesampleDataofobject. Additional parameters to thefeatureValuesfunction (that is used to extract the feature value matrix) can be passed via....
Visualization
plot(): plot for each file the position of individual peaks in the m/z - retention time space (with color-coded intensity) and a base peak chromatogram. This function should ideally be called only on a data subset (i.e. after usingfilterRt()andfilterMz()to restrict to a region of interest). ParametermsLevelallows to define from which MS level the plot should be created. Ifxis aXcmsExperimentwith available identified chromatographic peaks, also the region defining the peaks are indicated with a rectangle. ParameterpeakColallows to define the color of the border for these rectangles.plotAdjustedRtime(): plot the alignment results; seeplotAdjustedRtime()for more information.plotChromPeakImage(): show the density of identified chromatographic peaks per file along the retention time. SeeplotChromPeakImage()for details.plotChromPeaks(): indicate identified chromatographic peaks from one sample in the RT-m/z space. SeeplotChromPeaks()for details.
General functionality and functions for backward compatibility
uniqueMsLevels(): returns the unique MS levels of the spectra inobject.
The functions listed below ensure compatibility with the older
XCMSnExp() xcms result object. Also, an XcmsExperiment can be coerced
to the older XCMSnExp class using as(object, "XCMSnExp") same as a
XCMSnExp class can be coerced to XcmsExperiment using
as(object, "XcmsExperiment").
fileNames(): returns the original data file names for the spectra data. Ideally, thedataOriginordataStoragespectra variables from the object'sspectra()should be used instead.fromFile(): returns the file (sample) index for each spectrum withinobject. Generally, subsetting by sample using the[is the preferred way to get spectra from a specific sample.polarity(): returns the polarity information for each spectrum inobject.processHistory(): returns alistwith ProcessHistory process history objects that contain also the parameter object used for the different processings. Optional parametertypeallows to query for specific processing steps.rtime(): extract retention times of the spectra from theMsExperimentorXcmsExperimentobject. It is thus a shortcut forrtime(spectra(object))which would be the preferred way to extract retention times from anMsExperiment. Thertime()method forXcmsExperimenthas an additional parameteradjustedwhich allows to define whether adjusted retention times (if present -adjusted = TRUE) or raw retention times (adjusted = FALSE) should be returned. By default adjusted retention times are returned if available.
Differences compared to the XCMSnExp() object
Subsetting by
[supports arbitrary ordering.
Examples
## Create a MsExperiment object representing the data from an LC-MS
## experiment.
library(MsExperiment)
library(MsDataHub)
library(faahKO)
## Define the raw data files
fls <- c(system.file('cdf/KO/ko15.CDF', package = "faahKO"),
system.file('cdf/KO/ko16.CDF', package = "faahKO"),
system.file('cdf/KO/ko18.CDF', package = "faahKO"))
## Define a data frame with the sample characterization
df <- data.frame(mzML_file = basename(fls),
sample = c("ko15", "ko16", "ko18"))
## Importe the data. This will initialize a `Spectra` object representing
## the raw data and assign these to the individual samples.
mse <- readMsExperiment(spectraFiles = fls, sampleData = df)
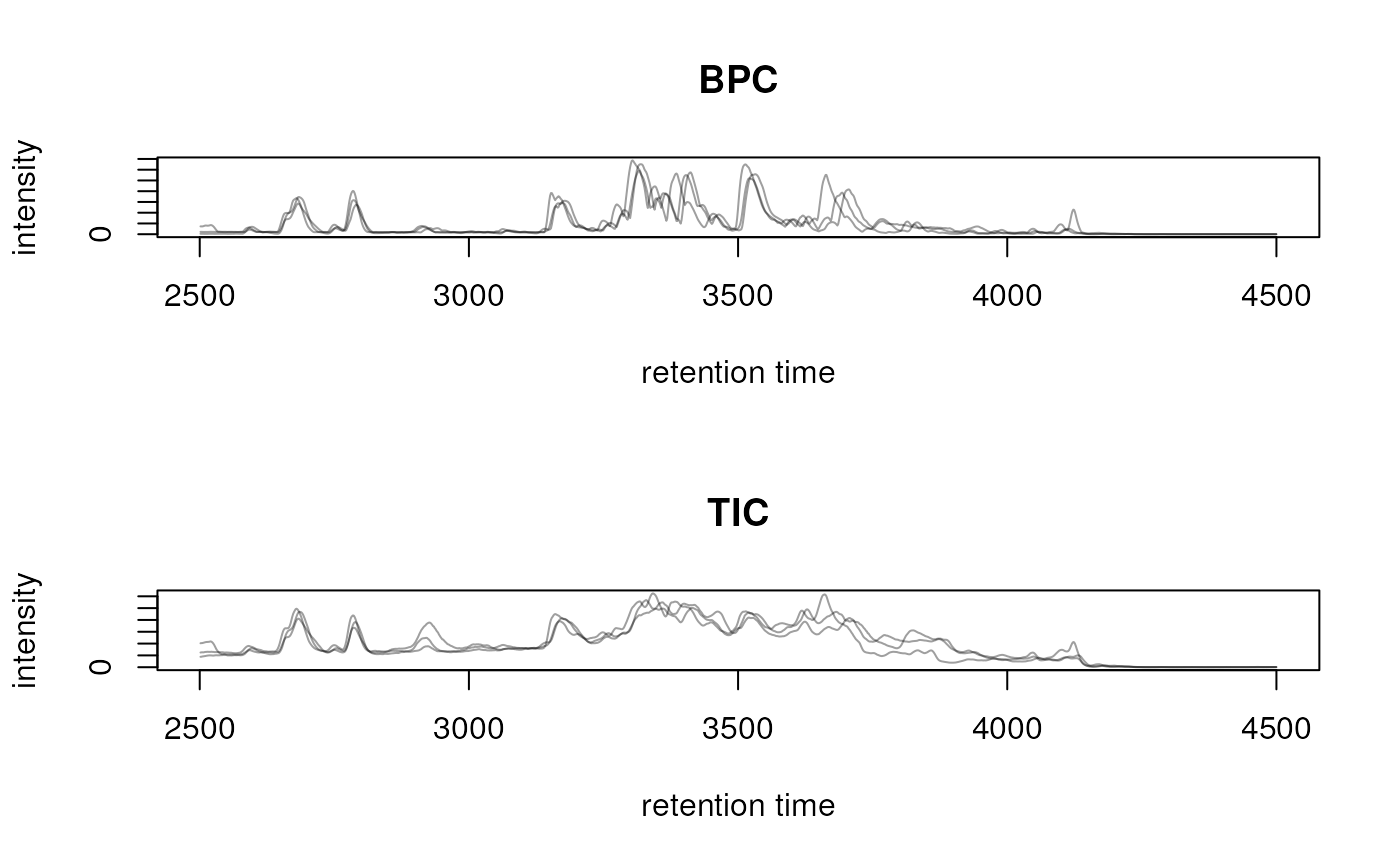
## Extract a total ion chromatogram and base peak chromatogram
## from the data
bpc <- chromatogram(mse, aggregationFun = "max")
tic <- chromatogram(mse)
## Plot them
par(mfrow = c(2, 1))
plot(bpc, main = "BPC")
plot(tic, main = "TIC")
 ## Extracting MS2 chromatographic data
##
## To show how MS2 chromatograms can be extracted we first load a DIA
## (SWATH) data set.
library(MsDataHub)
mse_dia <- readMsExperiment(PestMix1_SWATH.mzML())
#> see ?MsDataHub and browseVignettes('MsDataHub') for documentation
#> loading from cache
## Extracting MS2 chromatogram requires also to specify the isolation
## window from which to extract the data. Without that chromatograms
## will be empty:
chr_ms2 <- chromatogram(mse_dia, msLevel = 2L)
intensity(chr_ms2[[1L]])
#> [1] 167.451218 105.432610 48.448413 42.587666 51.141832
#> [6] 32.010572 39.833950 162.208892 146.962103 116.968997
#> [11] 47.464513 43.562779 47.522917 32.687743 48.205317
#> [16] 154.777987 151.650104 114.490964 51.955748 40.298667
#> [21] 42.889420 26.168091 41.573644 160.917377 155.598118
#> [26] 115.631891 56.064858 40.531724 51.336031 35.671689
#> [31] 47.007723 176.209493 150.000631 129.402529 52.351775
#> [36] 41.211416 45.764487 30.722598 42.549656 162.606472
#> [41] 159.749701 131.409168 57.591062 40.961098 47.379330
#> [46] 35.990142 56.194426 176.991264 158.162751 128.185092
#> [51] 55.115210 38.549212 52.187233 29.735383 42.294920
#> [56] 168.506534 148.606744 123.903268 50.443994 37.206104
#> [61] 49.667910 36.124219 45.099398 160.804559 144.212288
#> [66] 117.309985 57.318721 40.807128 46.997778 31.888547
#> [71] 44.914434 162.760728 141.112480 116.353180 61.330481
#> [76] 42.391655 46.165895 36.830656 41.048551 173.389121
#> [81] 150.354805 121.448249 58.297946 39.415169 48.137585
#> [86] 33.945039 42.597284 176.705435 152.759067 117.964302
#> [91] 57.009782 41.762080 49.519787 33.609967 45.789184
#> [96] 173.561615 131.868021 118.099061 55.553144 39.780311
#> [101] 46.840269 34.194347 41.879404 157.716579 140.589185
#> [106] 121.374668 51.687723 40.195854 47.690116 37.602680
#> [111] 43.965245 156.025829 157.369269 110.587061 52.018941
#> [116] 39.871676 47.531537 33.916085 39.490682 162.735606
#> [121] 151.371999 115.130247 52.206799 41.082679 48.981736
#> [126] 29.885040 50.262584 174.824692 137.808708 118.701492
#> [131] 52.714717 36.914359 44.841750 35.428872 42.102767
#> [136] 176.969381 143.918775 122.172542 46.627960 39.409697
#> [141] 45.892751 32.955673 45.916826 164.074529 110.974725
#> [146] 121.420453 57.000711 38.071610 43.426923 31.968076
#> [151] 43.205524 167.359458 136.644147 114.657853 53.570604
#> [156] 37.251428 48.855616 28.654548 46.821310 159.257533
#> [161] 136.078653 122.850526 51.119067 36.936050 47.987294
#> [166] 31.154454 44.033667 187.065329 138.560486 121.384740
#> [171] 49.859420 41.431116 52.683159 31.240847 41.930264
#> [176] 166.090714 137.037807 126.818365 55.785933 40.353991
#> [181] 47.617070 32.448793 46.101182 178.091599 126.628694
#> [186] 125.285128 53.154098 38.196966 45.725605 33.988735
#> [191] 47.783238 176.030519 138.156590 121.158414 51.131236
#> [196] 37.260215 46.927999 32.118103 50.824118 163.126561
#> [201] 143.490280 119.269498 50.446773 39.403250 45.444075
#> [206] 31.371791 46.041094 163.995802 98.558906 97.119329
#> [211] 43.657266 31.607119 40.415288 19.480071 41.681597
#> [216] 165.475850 101.389368 103.396216 42.729989 20.902430
#> [221] 48.426218 26.930463 53.396440 187.151969 58.083847
#> [226] 108.166753 51.910657 24.056782 43.929434 29.711441
#> [231] 46.996525 143.340123 111.724883 130.201873 46.946882
#> [236] 30.703287 45.463666 25.532886 53.455083 188.459844
#> [241] 121.595032 128.265759 48.481593 36.367438 39.432143
#> [246] 35.110147 45.317950 199.461130 123.822349 124.453397
#> [251] 51.373002 40.373744 44.575361 34.098693 44.992004
#> [256] 199.302711 115.622113 115.520087 52.588235 36.559259
#> [261] 42.863897 19.107814 42.420217 216.812980 145.216247
#> [266] 118.223439 61.237153 31.971820 45.693190 30.493408
#> [271] 43.479256 214.007843 137.507353 135.967656 60.011754
#> [276] 37.082835 39.620731 30.998244 42.027486 232.012743
#> [281] 118.387136 142.364895 51.672463 38.945555 41.669065
#> [286] 33.612390 41.247462 196.533657 115.020205 142.988161
#> [291] 46.530636 36.358999 43.610314 34.859240 41.568579
#> [296] 199.727458 118.619273 127.635654 47.808546 34.739862
#> [301] 45.326667 38.867514 43.806293 202.771954 126.833096
#> [306] 164.186773 52.443501 41.062466 45.812602 36.456879
#> [311] 41.972819 222.238004 129.047394 146.835109 53.216151
#> [316] 36.054789 41.644602 41.153462 44.151703 198.428224
#> [321] 133.110851 127.782179 50.909340 39.017341 43.719600
#> [326] 42.062764 44.099596 187.029552 123.340463 116.238022
#> [331] 46.525230 41.095261 49.200918 45.152890 47.486162
#> [336] 226.301902 134.037386 124.916782 58.381726 40.923225
#> [341] 49.689228 36.423063 44.652538 161.173165 126.906557
#> [346] 103.237367 55.496240 42.352044 45.500991 34.862405
#> [351] 41.926576 163.237384 117.727915 105.705640 49.150506
#> [356] 38.811569 48.849277 27.522183 44.785886 160.226922
#> [361] 113.355423 112.975909 50.373055 39.141868 47.454134
#> [366] 31.329878 46.244481 158.126581 121.931115 118.672109
#> [371] 56.822938 37.047153 44.821043 35.484114 38.755738
#> [376] 191.300899 130.326700 117.394953 53.562705 36.812964
#> [381] 45.737268 31.711265 43.831297 158.679602 128.274776
#> [386] 111.081271 48.823742 39.079050 45.954053 31.563248
#> [391] 38.653086 176.238025 128.692405 118.366005 42.698888
#> [396] 39.021745 46.105397 35.345455 40.248080 175.256934
#> [401] 139.035411 112.353086 50.371917 37.354196 48.254171
#> [406] 32.303338 42.679164 171.239180 104.472487 122.455740
#> [411] 48.074323 38.934960 46.869484 35.927003 45.715158
#> [416] 228.863116 130.873426 123.462988 56.414827 41.828009
#> [421] 52.009153 37.171067 46.885994 241.372911 123.043119
#> [426] 133.669185 61.397251 41.912858 51.326151 34.145175
#> [431] 43.159684 193.986114 112.600631 120.207558 41.959882
#> [436] 33.638388 47.956467 32.989986 38.815009 182.225208
#> [441] 110.543328 126.495590 45.878389 30.322753 40.737404
#> [446] 27.611870 45.191360 162.945866 108.900070 118.947459
#> [451] 45.752574 29.518900 43.446930 28.691413 40.876603
#> [456] 152.296499 97.336724 116.545222 50.060893 34.709605
#> [461] 41.048271 35.368648 39.523235 152.532517 97.537269
#> [466] 92.912022 45.620119 29.189715 36.658149 26.662089
#> [471] 38.752345 128.852280 100.981794 109.578946 48.040893
#> [476] 34.831760 38.101074 28.549529 41.152690 141.402519
#> [481] 101.452419 113.073400 50.223908 32.221343 41.203864
#> [486] 30.189605 41.476329 150.812408 119.987223 126.791050
#> [491] 56.253979 33.970643 41.440763 28.418729 36.884502
#> [496] 151.521866 127.808146 107.266956 50.975376 35.513003
#> [501] 42.469854 30.469824 36.575157 145.856516 112.498257
#> [506] 97.783282 52.686416 34.647375 40.969349 27.201655
#> [511] 39.861164 147.592851 125.196846 107.886744 51.600730
#> [516] 37.969832 37.686576 33.473837 40.492766 138.681531
#> [521] 115.332075 116.144761 47.054372 33.797785 46.507380
#> [526] 27.791961 42.171336 140.036967 121.225005 104.036724
#> [531] 47.233675 38.214997 41.466050 32.206779 42.691704
#> [536] 139.523492 129.458122 111.883703 45.873956 34.418329
#> [541] 39.452800 28.672196 44.544770 132.852484 105.496340
#> [546] 98.788578 49.043451 33.354703 37.570898 28.401155
#> [551] 46.793168 124.081183 134.779078 111.072208 48.193504
#> [556] 37.352554 38.492679 30.140132 34.180987 150.349048
#> [561] 127.751322 109.830612 48.353302 33.361524 38.676967
#> [566] 27.177009 38.237094 151.832900 119.423590 108.777089
#> [571] 59.370222 27.981177 41.502313 28.070891 33.805593
#> [576] 153.805769 66.500519 100.033210 38.243472 21.429552
#> [581] 35.849464 27.832320 42.276980 141.855202 108.842237
#> [586] 113.209048 40.932875 31.415275 26.385132 25.796249
#> [591] 44.596828 136.734716 39.113411 88.657524 43.650184
#> [596] 30.565965 44.374236 25.166760 39.799153 139.989898
#> [601] 112.345237 105.048307 47.806467 34.410027 33.377036
#> [606] 25.100477 33.934663 134.232939 109.667560 108.007680
#> [611] 46.322747 31.528887 36.890234 28.285215 32.815617
#> [616] 129.861855 103.835167 105.723162 46.497357 27.632753
#> [621] 39.557204 28.456075 37.007709 122.480471 91.297184
#> [626] 99.087357 41.633584 31.281372 38.874662 30.189158
#> [631] 38.613619 133.617479 102.807951 97.771981 48.130536
#> [636] 30.429434 35.423973 26.915430 40.223614 139.184034
#> [641] 106.234774 104.388371 48.370299 31.882032 34.893013
#> [646] 25.255039 36.265976 129.206981 101.493353 99.002509
#> [651] 44.346640 32.158756 39.429191 26.705643 37.622683
#> [656] 132.237249 109.599833 92.242120 46.847184 31.010174
#> [661] 37.535520 27.512162 41.985617 137.100186 105.086888
#> [666] 94.188571 43.261979 33.923542 40.955904 24.140754
#> [671] 49.249031 145.383441 108.475989 102.639441 44.052413
#> [676] 31.671950 38.316922 26.313719 36.032487 145.339185
#> [681] 109.010582 111.256913 44.568090 32.622182 43.235431
#> [686] 28.846874 41.603560 185.257926 90.491924 55.381179
#> [691] 52.559675 35.404760 39.212934 26.798098 37.332781
#> [696] 207.062895 113.098940 112.052358 46.283524 25.168956
#> [701] 36.233487 27.672381 44.556306 179.957265 110.232423
#> [706] 102.707046 48.953326 36.362766 35.710726 34.303696
#> [711] 39.764515 188.588142 113.188109 118.948407 48.587346
#> [716] 33.652624 40.238380 31.320930 29.659764 191.976195
#> [721] 104.352510 105.337010 47.377342 30.138890 35.353116
#> [726] 30.443518 38.085864 161.939634 99.286794 113.933793
#> [731] 47.386050 31.276776 35.586038 28.099018 38.582196
#> [736] 158.502928 108.189300 108.556956 45.916943 29.664124
#> [741] 35.072582 29.660793 30.060255 163.596847 104.458447
#> [746] 111.339333 42.839141 30.381518 32.467032 27.207105
#> [751] 31.829775 152.395823 92.090382 101.389516 38.136271
#> [756] 30.162735 34.126044 27.465397 32.316456 159.614114
#> [761] 97.103220 93.203515 44.641377 27.876265 36.166051
#> [766] 25.380971 33.702906 155.271182 102.884171 96.148820
#> [771] 44.848030 30.642579 37.470218 27.501099 30.589302
#> [776] 163.899956 101.194633 95.039853 45.704995 32.728440
#> [781] 33.720749 26.186973 36.896967 156.103823 113.336619
#> [786] 98.596145 48.631773 33.012071 38.892305 28.817390
#> [791] 39.441818 171.890864 112.334461 107.901088 46.708054
#> [796] 32.391567 29.360276 26.269499 32.755082 160.337011
#> [801] 113.112331 96.390912 47.874924 32.144085 34.075555
#> [806] 32.297730 35.310021 149.723302 104.396845 99.148335
#> [811] 44.486707 31.296760 33.825723 29.673124 34.026282
#> [816] 165.011075 109.287485 105.432956 46.554563 31.546334
#> [821] 34.769495 26.309489 36.822224 170.078539 97.233150
#> [826] 103.437863 43.135479 28.860388 33.053009 23.208648
#> [831] 38.422893 159.336934 88.512512 103.206401 47.365989
#> [836] 31.572353 35.984752 22.722223 33.728057 146.322609
#> [841] 107.862552 105.438540 47.479481 28.481425 31.647929
#> [846] 26.164322 30.565289 149.787435 130.332740 98.296281
#> [851] 41.290008 38.151850 34.086264 29.415540 37.504760
#> [856] 201.926000 210.496647 93.751699 50.988839 51.721273
#> [861] 47.046052 29.129116 59.443360 349.692621 411.374657
#> [866] 85.913687 59.676289 90.024034 67.344532 40.821380
#> [871] 91.276277 530.047933 697.728942 79.454606 74.053664
#> [876] 132.664091 82.780161 49.933027 132.319325 649.397279
#> [881] 841.452977 91.543068 92.976836 168.231806 102.184256
#> [886] 58.848030 135.167703 733.247639 900.496888 94.106439
#> [891] 90.594612 144.141867 95.562042 51.211730 137.975992
#> [896] 645.368867 897.487709 86.226899 88.836395 139.545482
#> [901] 74.780693 50.911947 104.015507 608.068443 704.566139
#> [906] 94.131264 68.269341 92.880510 63.487855 35.247855
#> [911] 72.120525 399.302359 426.784746 87.379760 60.358757
#> [916] 63.452186 54.776193 29.311635 52.562169 282.731257
#> [921] 278.563776 96.429320 49.760013 48.450612 38.933191
#> [926] 25.844857 43.225975 212.039464 160.201024 98.777070
#> [931] 46.029153 37.862905 37.250173 27.008984 35.842104
#> [936] 169.997601 156.630668 93.014267 48.222163 35.064183
#> [941] 35.451472 23.543123 32.686649 171.065548 105.790041
#> [946] 108.830875 45.272460 31.981356 30.120026 23.585034
#> [951] 32.319106 159.294988 115.495441 115.896828 47.765108
#> [956] 29.215978 34.400212 25.209040 35.119547 165.818130
#> [961] 107.669547 106.891347 46.208358 32.254054 33.814447
#> [966] 24.801176 35.543628 155.108299 113.496329 110.971571
#> [971] 44.721446 28.139904 31.768555 25.935012 31.160421
#> [976] 141.595916 104.123449 109.959406 42.095975 29.355523
#> [981] 33.334306 28.015602 32.062996 150.583101 90.077886
#> [986] 105.341658 41.192123 29.251126 29.782968 26.674842
#> [991] 36.906096 170.581104 97.567593 105.959960 39.480080
#> [996] 28.286513 28.900908 22.996521 31.173936 142.981469
#> [1001] 93.127804 109.395854 44.881067 30.412322 33.138231
#> [1006] 24.754524 35.953917 140.121663 102.959301 105.862271
#> [1011] 40.174324 29.285178 32.617315 27.309924 38.589327
#> [1016] 166.073498 89.320133 106.740038 46.417630 29.564379
#> [1021] 39.726060 29.746300 34.125606 168.017869 90.316623
#> [1026] 107.312059 42.062266 29.621464 34.036371 26.995349
#> [1031] 34.642726 157.013142 97.627014 114.828925 46.800743
#> [1036] 27.051983 35.225135 28.283421 35.625325 175.540814
#> [1041] 118.424921 107.035100 43.272736 30.311741 34.189589
#> [1046] 27.176501 34.648530 180.816095 107.999549 106.611958
#> [1051] 44.284883 29.429024 34.550493 26.800240 36.931911
#> [1056] 156.397981 102.388683 114.543137 43.335283 29.670092
#> [1061] 36.463383 31.204052 36.964631 152.760545 105.690832
#> [1066] 112.532788 45.675366 31.084984 36.516981 27.889156
#> [1071] 39.951343 160.542475 108.768802 120.086017 44.621606
#> [1076] 29.979912 32.758499 29.640258 35.449074 144.627534
#> [1081] 95.904708 98.581966 29.151899 25.652984 34.015049
#> [1086] 23.739257 33.309044 132.845364 110.519425 97.435606
#> [1091] 36.445994 24.191037 33.755435 25.317860 34.692138
#> [1096] 133.370595 89.432865 78.788986 35.394559 32.745781
#> [1101] 34.401351 21.857643 37.652579 149.387315 92.966335
#> [1106] 117.034547 41.307684 25.799113 35.371046 30.828860
#> [1111] 36.504718 148.164662 173.861551 98.670328 50.817050
#> [1116] 24.953204 36.405526 20.948719 52.882848 151.105567
#> [1121] 391.764819 158.707810 32.949967 33.791382 46.733799
#> [1126] 21.774906 87.747553 160.094614 819.146131 141.687122
#> [1131] 41.526904 44.907794 62.191336 38.025100 108.899176
#> [1136] 242.205228 746.278612 165.371112 43.624310 52.703623
#> [1141] 95.556257 46.402372 204.718860 409.282334 1610.046856
#> [1146] 268.615342 60.597154 55.776300 92.681383 48.520625
#> [1151] 204.365289 371.091568 1585.529313 261.537745 35.539510
#> [1156] 62.214589 96.423575 29.725105 188.243389 314.450925
#> [1161] 1120.563718 255.135276 47.119980 48.516808 89.928485
#> [1166] 37.324552 142.475128 250.435000 566.727559 168.645876
#> [1171] 40.371340 44.392902 66.118259 29.763397 90.735531
#> [1176] 191.261879 789.235621 177.715166 49.942617 39.819652
#> [1181] 46.928773 29.820479 76.079930 172.221738 431.544954
#> [1186] 142.811706 44.438606 33.643979 39.060616 25.329606
#> [1191] 55.605822 151.479111 287.372645 130.826744 39.162669
#> [1196] 31.567921 36.137109 25.640328 36.565181 156.137129
#> [1201] 189.563114 114.160943 48.205198 27.381770 30.582241
#> [1206] 22.257580 40.508673 146.431932 116.312190 100.976158
#> [1211] 36.645808 27.605937 30.085477 26.043214 36.610432
#> [1216] 184.984871 126.509686 90.825038 36.832579 27.637315
#> [1221] 32.743402 25.398717 30.508300 140.194227 118.434799
#> [1226] 95.947858 29.674962 28.870998 33.375377 27.328116
#> [1231] 39.398779 178.195530 105.098561 84.987955 43.518142
#> [1236] 27.896133 36.033127 27.324011 34.533595 153.969597
#> [1241] 85.703834 99.892381 34.242776 26.875551 36.100405
#> [1246] 23.063659 30.791688 165.702633 67.073723 119.794516
#> [1251] 39.406710 22.804362 32.508115 31.149893 38.454433
#> [1256] 139.124003 99.513846 77.327728 43.546272 31.916123
#> [1261] 33.064523 31.806988 37.525104 183.940540 132.497918
#> [1266] 99.188331 47.054355 30.830371 34.161061 27.230252
#> [1271] 39.881592 186.165895 84.479401 96.370104 44.697988
#> [1276] 28.816349 31.995093 32.431530 39.123189 152.214855
#> [1281] 63.328493 124.861869 41.193206 28.866022 40.435678
#> [1286] 25.878933 35.254059 126.227156 110.655427 115.221361
#> [1291] 35.455676 30.530741 37.684927 17.316758 32.490885
#> [1296] 134.756237 105.090005 141.773908 34.771503 30.138122
#> [1301] 29.993710 19.134206 31.945019 174.943336 86.977711
#> [1306] 119.414188 46.054079 27.007063 37.394333 25.526424
#> [1311] 32.350547 176.360991 100.247268 92.214134 42.100218
#> [1316] 26.598997 31.552884 32.203519 33.252210 104.017945
#> [1321] 116.210076 62.106982 49.084530 26.358952 34.067227
#> [1326] 26.564477 36.290081 142.263964 47.620735 112.944846
#> [1331] 34.322501 28.551036 38.993419 22.362873 35.850753
#> [1336] 136.374939 99.798530 92.174216 24.876948 31.416041
#> [1341] 27.977401 22.599160 27.002678 136.757612 85.021712
#> [1346] 66.102933 48.928459 27.674189 31.963448 31.680869
#> [1351] 31.415532 206.067120 79.858983 67.724770 51.565559
#> [1356] 36.770206 33.478235 33.397290 30.300532 185.169355
#> [1361] 59.689057 113.624347 45.834682 35.071004 30.272516
#> [1366] 26.018613 31.311628 137.484421 97.771376 98.282888
#> [1371] 46.533382 32.927842 31.117646 27.572587 33.224553
#> [1376] 150.592315 72.236452 96.214203 52.651345 36.777134
#> [1381] 28.324229 32.304347 38.065710 116.620451 98.368924
#> [1386] 106.193656 54.976777 26.129837 31.190081 29.061710
#> [1391] 33.049347 235.458633 49.644926 105.635875 28.457881
#> [1396] 32.916288 41.376448 28.199523 32.091286 204.683036
#> [1401] 92.369982 117.488798 33.176475 37.188275 36.916034
#> [1406] 24.631105 35.289954 152.838308 74.815710 74.688163
#> [1411] 56.601092 27.957361 35.494486 34.825380 37.205302
#> [1416] 162.042605 49.309894 107.588334 42.188854 29.120570
#> [1421] 35.488853 19.608062 31.033799 186.844817 97.331014
#> [1426] 93.206562 48.051964 37.413884 27.915677 40.912424
#> [1431] 38.188606 119.108399 53.634031 105.031201 43.672971
#> [1436] 21.503384 31.892517 27.013946 45.004002 110.739233
#> [1441] 95.821644 93.103918 58.389068 28.267440 35.444377
#> [1446] 24.704700 36.539813 212.861125 103.871532 129.800170
#> [1451] 40.755682 30.372291 37.629196 28.002186 35.415052
#> [1456] 235.109370 80.404990 123.798571 36.751008 41.045761
#> [1461] 29.878926 24.239642 40.312472 127.568099 57.314662
#> [1466] 97.914116 46.504641 29.215859 38.111446 25.698333
#> [1471] 34.554269 157.990302 84.496738 74.541171 32.664953
#> [1476] 40.293004 28.729528 28.841443 38.652989 144.608685
#> [1481] 103.610632 91.145180 38.251578 32.525705 29.559268
#> [1486] 28.371520 34.002482 139.517177 47.311838 122.164686
#> [1491] 46.971462 28.612430 36.331883 27.220920 31.552084
#> [1496] 207.302292 91.903460 114.640190 30.747115 26.480455
#> [1501] 37.264549 30.834890 32.124568 183.139218 70.849133
#> [1506] 129.735962 40.666440 24.421463 35.638740 17.057095
#> [1511] 35.863203 160.275320 89.107102 123.293859 35.963508
#> [1516] 39.949520 35.584113 20.181869 31.828364 135.130744
#> [1521] 119.125820 96.000743 45.748627 25.493576 37.545546
#> [1526] 21.838315 35.173620 172.009139 70.488143 104.181722
#> [1531] 25.083607 32.690491 41.949165 15.853308 37.556713
#> [1536] 156.180366 89.693465 88.501997 23.182854 33.387140
#> [1541] 39.341711 19.210659 38.406144 160.599245 123.616225
#> [1546] 92.213588 36.325025 32.144818 46.369477 39.971955
#> [1551] 33.936432 195.647481 77.179203 111.316890 33.769247
#> [1556] 26.788419 46.454249 24.763817 39.588913 146.920034
#> [1561] 126.582540 109.726625 40.927068 38.069900 41.180900
#> [1566] 27.001062 34.850588 197.805155 80.765977 97.622657
#> [1571] 26.326894 27.408477 42.529164 21.699734 30.852826
#> [1576] 171.079778 93.319155 103.618405 31.096392 37.420658
#> [1581] 33.034041 21.859346 35.250289 171.183623 87.360047
#> [1586] 100.242504 32.113428 26.525980 42.798271 20.262626
#> [1591] 28.644451 176.150627 69.840818 119.224692 25.607179
#> [1596] 31.822188 35.727435 21.551686 35.178318 156.434354
#> [1601] 104.369524 101.711197 53.031272 29.573143 32.794880
#> [1606] 29.026631 36.193032 154.152449 89.099312 98.262950
#> [1611] 40.983253 24.163189 34.501748 31.976277 35.426385
#> [1616] 148.537631 97.701749 109.391669 46.714138 25.523892
#> [1621] 31.964336 35.362196 33.886820 157.480453 86.366383
#> [1626] 79.107788 38.764546 26.201341 25.624606 28.607790
#> [1631] 33.253877 142.411533 90.983309 93.451915 47.661561
#> [1636] 26.865360 32.096030 31.834129 31.258656 230.045142
#> [1641] 71.557235 132.132516 47.257443 24.495178 37.002452
#> [1646] 20.361930 36.194230 248.565752 64.068487 157.677227
#> [1651] 53.574143 24.249650 37.743398 21.712505 28.686528
#> [1656] 218.324965 66.007129 89.803788 46.140171 23.027989
#> [1661] 29.306930 27.006135 32.963365 187.819705 85.063270
#> [1666] 81.274325 43.366147 28.403436 37.554051 21.598555
#> [1671] 36.212499 158.448519 78.372012 77.477039 29.996444
#> [1676] 32.272852 32.906730 24.015367 33.172171 272.487378
#> [1681] 84.846888 85.014031 29.668608 39.581763 40.042868
#> [1686] 37.099377 36.948299 203.872117 61.969520 146.510894
#> [1691] 38.981498 30.018676 20.476881 36.929195 43.787444
#> [1696] 154.430081 90.605106 146.302234 40.669078 27.420887
#> [1701] 35.429644 57.348454 34.920677 263.241275 71.016427
#> [1706] 79.633341 50.394459 25.392463 40.220837 52.619259
#> [1711] 42.117045 215.196783 58.495471 108.490964 61.623339
#> [1716] 23.415285 38.055228 26.456648 61.001604 266.378869
#> [1721] 93.817655 121.125686 57.490407 37.148676 27.849703
#> [1726] 45.148767 50.306234 197.005061 106.544321 102.487807
#> [1731] 27.663152 38.055031 43.434172 48.045937 40.335960
#> [1736] 191.640031 67.029701 137.051483 65.579437 22.572407
#> [1741] 38.649261 40.508534 57.055165 140.799223 133.669713
#> [1746] 74.303656 56.998198 39.294406 36.815819 25.600391
#> [1751] 54.495908 295.211768 127.769307 109.169560 30.880279
#> [1756] 32.020910 43.418441 48.313316 48.187284 147.377280
#> [1761] 94.739802 119.769936 46.917040 39.250639 41.261021
#> [1766] 28.507046 41.979571 196.337320 81.106436 168.109136
#> [1771] 52.962343 21.686875 30.963254 43.027813 38.210871
#> [1776] 307.503439 80.468017 141.685174 47.534258 29.558209
#> [1781] 37.009343 37.449933 41.153272 166.665936 105.003535
#> [1786] 86.877174 55.994831 38.115102 45.375383 23.711647
#> [1791] 45.017327 195.095036 64.489254 141.102600 58.586026
#> [1796] 24.740041 39.417257 45.935621 44.368479 248.729436
#> [1801] 99.076422 108.528579 57.070673 37.177808 25.617253
#> [1806] 43.001979 56.238403 242.362830 68.406010 149.126453
#> [1811] 26.213064 36.171577 40.075221 32.053932 54.676368
#> [1816] 356.667826 55.901124 155.707960 67.176312 30.555825
#> [1821] 36.290777 51.125159 69.950411 172.199389 84.890150
#> [1826] 155.172876 20.486742 34.701371 45.691073 48.161121
#> [1831] 36.894187 302.110497 93.596723 91.851749 65.116098
#> [1836] 39.147736 36.466925 26.774093 69.335105 296.318700
#> [1841] 90.782109 215.716420 51.973785 27.257805 30.972503
#> [1846] 49.214708 58.873692 155.052388 131.714048 176.217341
#> [1851] 27.778185 31.060168 43.479056 37.612360 33.331283
#> [1856] 271.083706 108.032408 101.957330 52.877356 36.519604
#> [1861] 33.477509 23.041509 56.101345 305.169499 113.602917
#> [1866] 211.426262 53.749557 18.842347 39.586898 44.027395
#> [1871] 43.585771 185.544784 99.435757 98.333853 53.386409
#> [1876] 34.430626 30.946110 25.427338 38.509870 285.530040
#> [1881] 52.472315 198.993257 58.660556 38.832734 37.082364
#> [1886] 44.011392 41.031400 188.437410 87.474563 98.043931
#> [1891] 78.003673 34.698912 41.750663 35.420446 39.510279
#> [1896] 383.074116 98.765139 119.145008 51.552674 33.715571
#> [1901] 34.694111 42.824697 48.457643 296.863562 84.082733
#> [1906] 114.538239 28.738582 42.499469 43.984079 36.904957
#> [1911] 35.895894 325.381169 81.027964 137.467786 38.488739
#> [1916] 27.329626 47.504072 16.035063 58.288272 280.982411
#> [1921] 81.964843 147.251262 41.919116 28.917983 29.126941
#> [1926] 24.707881 54.181098 242.407159 100.310015 137.655731
#> [1931] 41.494815 26.177019 28.914556 26.650264 46.273476
#> [1936] 231.882754 89.895481 112.320566 32.803755 33.588569
#> [1941] 33.033767 26.608555 50.960213 458.659131 107.640478
#> [1946] 150.360576 65.517246 35.414668 38.215521 43.125685
#> [1951] 50.555097 283.978981 97.550146 135.993181 58.664513
#> [1956] 35.917116 36.995451 38.546234 49.043232 325.272207
#> [1961] 98.996154 147.221397 69.319522 35.468042 35.986838
#> [1966] 38.794882 50.329541 335.374756 96.065313 147.845948
#> [1971] 61.414690 35.989945 36.926313 38.591692 51.843903
#> [1976] 324.264422 101.529915 135.551127 65.209398 36.824668
#> [1981] 37.633576 39.007057 46.347952 316.556611 96.712077
#> [1986] 142.219890 66.475186 34.892562 35.069106 41.596664
#> [1991] 52.278679 254.675998 109.176872 154.631251 65.022175
#> [1996] 43.055877 43.341532 49.361935 54.482969 354.457471
#> [2001] 89.377830 160.112253 64.213553 42.558553 39.835207
#> [2006] 59.856009 60.354117 379.372871 109.208437 166.123293
#> [2011] 61.810196 39.636466 40.870663 51.759527 54.428493
#> [2016] 376.155093 103.506800 170.954710 64.387163 40.418625
#> [2021] 39.887188 55.752389 56.483176 362.650536 115.075388
#> [2026] 143.364995 57.492267 38.523954 32.647061 51.301592
#> [2031] 46.871748 384.224012 88.074090 163.403510 47.091308
#> [2036] 38.166879 36.352915 38.096904 53.610835 368.810280
#> [2041] 108.832158 153.158567 67.981493 44.443709 39.159903
#> [2046] 52.367430 53.014402 415.838201 115.424278 153.048996
#> [2051] 120.144144 40.145176 36.078019 44.489095 52.712724
#> [2056] 389.350239 110.070706 146.372008 163.313465 46.588510
#> [2061] 40.543530 47.885738 53.107820 399.845750 96.952821
#> [2066] 155.735393 141.124334 46.514724 44.535562 46.303457
#> [2071] 52.881468 407.918184 114.145907 158.973029 121.196010
#> [2076] 32.334025 36.807087 48.935408 55.988427 400.204529
#> [2081] 122.143079 148.332373 109.835709 40.129369 39.771837
#> [2086] 36.824987 59.225614 334.339159 105.224713 149.474562
#> [2091] 79.284124 46.528001 41.737876 45.739151 70.237961
#> [2096] 403.477595 117.842087 495.825624 74.954894 34.224355
#> [2101] 40.451839 46.355804 62.324136 286.454247 89.056594
#> [2106] 5832.395161 87.522507 51.302731 41.023700 29.757374
#> [2111] 48.602331 235.819508 67.292766 13873.384850 112.370406
#> [2116] 75.664919 37.635591 27.751099 37.371573 267.709799
#> [2121] 52.310421 17165.405181 191.168944 82.199662 32.221482
#> [2126] 24.518181 33.193361 252.173733 49.401413 16437.503614
#> [2131] 159.827322 89.323235 36.391296 22.471497 35.772829
#> [2136] 252.290946 65.733328 13755.012642 97.936771 60.497118
#> [2141] 33.123704 27.679177 34.807008 253.211050 56.774516
#> [2146] 10955.394431 96.865632 51.837906 29.911206 29.050536
#> [2151] 40.047838 234.478684 82.087474 9493.409165 83.231644
#> [2156] 51.672872 35.252748 29.158378 48.748037 365.643260
#> [2161] 90.750294 6442.331874 75.831233 52.951960 38.056488
#> [2166] 33.368845 48.303094 352.849221 91.706198 4600.186775
#> [2171] 69.476735 52.822776 38.611727 44.488193 51.911443
#> [2176] 431.401173 101.796074 2892.800990 66.987991 51.283043
#> [2181] 38.911728 45.438261 47.577080 399.170704 108.112526
#> [2186] 1774.720067 64.928496 46.699510 43.879691 40.686856
#> [2191] 50.936232 445.515064 106.386173 1354.003510 64.540338
#> [2196] 49.147460 35.080291 46.991590 48.374540 428.179441
#> [2201] 103.169871 941.571921 72.048556 53.248166 36.666954
#> [2206] 48.456305 46.269972 422.903522 110.674410 752.972866
#> [2211] 70.220963 49.831384 37.652212 45.846990 49.459702
#> [2216] 447.458198 106.774150 630.149374 69.259278 51.613374
#> [2221] 33.897051 46.393051 46.725414 449.146344 97.630969
#> [2226] 541.745739 75.886827 41.253986 40.926556 45.904882
#> [2231] 45.775102 470.359972 99.460572 494.868960 72.884706
#> [2236] 52.137230 42.584389 46.582839 49.957400 438.727083
#> [2241] 110.900816 455.857940 75.793757 50.861001 43.644965
#> [2246] 48.120407 47.968101 472.913455 113.232404 394.845602
#> [2251] 73.335230 53.254563 39.055537 43.107564 44.777950
#> [2256] 486.724239 112.924868 342.480613 71.166023 52.477110
#> [2261] 43.093652 48.508074 47.582490 473.236068 107.121591
#> [2266] 314.538700 68.552210 49.572075 46.860595 42.282366
#> [2271] 41.354109 435.768428 106.979632 279.561779 63.373882
#> [2276] 53.443520 39.888940 47.507710 46.408509 479.389012
#> [2281] 99.470195 295.953485 70.021186 46.548519 40.014569
#> [2286] 43.371476 40.823099 454.627431 116.416887 241.948283
#> [2291] 65.018561 48.924704 37.893611 42.659015 49.039957
#> [2296] 491.670790 95.426186 237.162205 68.319898 53.581855
#> [2301] 36.353723 44.225962 45.001981 447.547469 109.786524
#> [2306] 237.586439 66.047106 45.552718 36.644631 43.799834
#> [2311] 44.847455 418.299828 122.267204 222.628184 64.904554
#> [2316] 46.077932 36.239206 50.909309 45.872858 475.359280
#> [2321] 121.625129 213.582292 64.780518 46.055814 41.639253
#> [2326] 43.247804 47.021251 482.833683 143.198061 203.076683
#> [2331] 61.808888 44.536787 41.674611 50.834753 49.958632
#> [2336] 462.927389 133.091117 212.156884 65.674832 45.832769
#> [2341] 45.227610 50.943171 45.399300 476.837618 134.462510
#> [2346] 212.997852 83.683738 46.307428 44.114147 49.426245
#> [2351] 46.066511 445.476788 146.815803 205.989675 147.752024
#> [2356] 50.177558 52.419657 55.526217 47.264174 490.760576
#> [2361] 170.817562 214.155841 181.371703 57.437563 52.451408
#> [2366] 60.217505 50.999636 485.747137 160.952902 195.593397
#> [2371] 186.363268 62.290553 47.320707 57.157110 49.019309
#> [2376] 485.676005 143.149765 199.440368 135.085678 76.823418
#> [2381] 45.874357 46.694395 46.705219 472.472557 122.590519
#> [2386] 196.164505 114.501037 81.142977 42.737055 47.379126
#> [2391] 43.960697 469.841693 120.431267 179.920177 88.308295
#> [2396] 75.505423 40.908099 55.427963 50.543494 447.686020
#> [2401] 112.830110 183.489394 70.699660 60.818758 41.392392
#> [2406] 63.049013 48.161211 447.263685 113.098922 186.604226
#> [2411] 70.344673 56.597906 43.191166 63.650399 43.669881
#> [2416] 478.137230 111.280911 183.160843 68.099514 54.380999
#> [2421] 45.587965 63.487612 47.277363 435.605030 113.039216
#> [2426] 163.528049 64.942955 46.626150 40.701930 53.573868
#> [2431] 44.855228 428.627071 111.044178 183.114925 71.578556
#> [2436] 40.439535 44.389309 57.025407 46.306825 438.637845
#> [2441] 106.830294 180.154460 71.048972 44.054351 41.686204
#> [2446] 47.594233 50.640354 477.948657 107.768497 183.448420
#> [2451] 70.589348 41.367258 40.588047 57.552715 43.678015
#> [2456] 505.276160 106.934839 175.443230 64.234972 45.826434
#> [2461] 41.444938 51.237273 49.127882 456.103902 110.304658
#> [2466] 181.128785 64.361756 42.257462 36.234890 56.042026
#> [2471] 46.002784 461.831220 112.112989 177.799746 59.419955
#> [2476] 45.835058 37.143329 57.791545 46.574726 473.481790
#> [2481] 113.347008 187.479618 60.441204 44.384575 36.748130
#> [2486] 58.255789 50.525020 468.894201 112.097804 172.268805
#> [2491] 66.881132 47.767594 35.614725 62.727494 47.721740
#> [2496] 479.832255 115.216485 168.147780 64.495323 48.895200
#> [2501] 40.707435 62.435092 44.537999 477.044890 107.602994
#> [2506] 160.913590 67.899740 48.596475 37.600284 50.072798
#> [2511] 44.891404 446.804062 111.290585 162.397966 61.712172
#> [2516] 41.225893 35.227686 51.303568 45.314574 461.196746
#> [2521] 115.938279 177.651151 67.345495 46.733009 39.527198
#> [2526] 56.816790 52.750451 424.494314 111.871450 163.096121
#> [2531] 80.499247 49.625922 40.675166 50.999692 52.492706
#> [2536] 468.263938 113.744053 176.055005 122.768205 47.463385
#> [2541] 41.041710 53.205646 78.578701 584.468115 115.955268
#> [2546] 191.130047 181.661404 54.624808 37.018977 56.041601
#> [2551] 70.841264 664.795500 124.590019 224.475239 187.583307
#> [2556] 50.438938 40.283084 50.180244 86.135610 570.187038
#> [2561] 134.230020 219.730538 146.569392 50.101408 39.866188
#> [2566] 49.076670 72.141985 521.389886 117.411897 200.841469
#> [2571] 111.737539 50.180836 39.118886 49.177277 63.998615
#> [2576] 466.357814 130.208352 186.026854 90.567113 47.000837
#> [2581] 39.512403 47.244710 59.240777 451.402441 121.219202
#> [2586] 183.565928 85.706048 51.367771 41.658139 46.595095
#> [2591] 62.208513 461.008189 112.948330 208.372259 106.603505
#> [2596] 50.952128 40.728191 55.395050 68.092937 587.144867
#> [2601] 121.209110 245.040250 166.608778 50.820778 46.078316
#> [2606] 54.586626 73.634932 629.747347 106.906315 220.482740
#> [2611] 174.262815 48.301433 46.288178 55.616089 77.046168
#> [2616] 610.995708 103.686176 194.916945 156.926065 48.564475
#> [2621] 44.978622 52.375992 70.770942 566.716943 114.624499
#> [2626] 180.656253 109.721550 47.479018 45.371014 50.931045
#> [2631] 70.955909 522.202017 110.237149 173.128564 100.635447
#> [2636] 45.183336 41.858009 48.651565 64.857451 435.670086
#> [2641] 109.835674 173.932545 76.596251 49.585715 44.332319
#> [2646] 47.835264 54.787442 399.844011 105.368428 158.937927
#> [2651] 71.319149 45.394350 41.681597 52.281926 49.528685
#> [2656] 382.456539 105.509972 162.843921 77.230597 44.006242
#> [2661] 37.321572 83.274316 49.168353 432.168623 105.166536
#> [2666] 154.536989 63.360656 43.257950 38.976037 121.911508
#> [2671] 41.409602 416.626463 104.332561 150.248674 65.020749
#> [2676] 48.399111 35.372174 97.019543 41.577903 386.252188
#> [2681] 99.126223 136.614468 69.607545 41.745846 33.963000
#> [2686] 89.715885 47.772692 373.844217 108.473919 138.761989
#> [2691] 107.148913 45.289243 38.850900 66.693888 47.320003
#> [2696] 374.427710 100.337538 147.019071 108.946940 44.767658
#> [2701] 31.970107 55.714322 40.898479 384.554854 104.902908
#> [2706] 152.970046 102.968200 42.386481 35.469126 46.910892
#> [2711] 39.892412 392.099680 111.670846 158.119160 93.352925
#> [2716] 43.845470 34.770151 43.459377 45.528240 368.951429
#> [2721] 100.894014 141.036437 91.908482 42.468380 35.463312
#> [2726] 43.798070 53.840996 376.266252 99.509117 133.894132
#> [2731] 89.294837 39.262945 37.729908 43.829460 216.280428
#> [2736] 351.838721 101.478866 140.720373 82.380093 40.751900
#> [2741] 37.179003 47.645561 397.230075 362.989834 98.861373
#> [2746] 140.412077 70.692329 50.636567 35.765085 50.151279
#> [2751] 320.469480 356.958652 108.716669 141.060782 78.796086
#> [2756] 43.260207 37.810845 44.797668 278.136920 334.220326
#> [2761] 93.462171 133.870650 69.984913 42.031692 39.729968
#> [2766] 42.617221 203.946074 375.335101 92.918614 150.667486
#> [2771] 66.658469 64.518478 40.465141 37.987780 319.792309
#> [2776] 381.687629 95.203955 158.715536 62.906516 118.817601
#> [2781] 35.928716 44.179380 422.806755 339.826894 96.870755
#> [2786] 170.204776 65.174303 129.831309 39.115973 46.567989
#> [2791] 277.553212 379.195222 100.939231 160.881076 61.380293
#> [2796] 113.236567 30.495809 43.721927 221.316362 331.970851
#> [2801] 101.783902 156.075804 65.746408 94.056235 33.463657
#> [2806] 41.203415 134.397904 335.178189 109.343455 159.628160
#> [2811] 56.701886 75.474413 33.982118 40.438525 92.386664
#> [2816] 413.892579 107.377343 207.107832 58.226374 60.382087
#> [2821] 39.293829 41.553251 67.557693 502.811196 100.060179
#> [2826] 241.096155 63.202887 55.581887 32.281917 48.897676
#> [2831] 54.018214 530.671079 98.657496 250.577511 64.236887
#> [2836] 57.254045 29.696300 53.560764 47.510936 482.104900
#> [2841] 87.972094 243.426023 68.539853 80.170418 33.892214
#> [2846] 60.884106 52.454029 417.665537 95.028840 202.186937
#> [2851] 65.538071 96.851122 35.226911 49.554699 47.346519
#> [2856] 452.073756 105.329216 179.652847 63.027096 89.270784
#> [2861] 32.109190 50.222711 40.365956 390.430038 94.825371
#> [2866] 154.653021 65.312407 74.879131 39.632950 43.034229
#> [2871] 44.376038 364.625164 106.327953 155.579487 56.476849
#> [2876] 68.204701 37.557180 40.732334 51.897565 338.730040
#> [2881] 101.356362 152.716527 56.022610 64.241037 38.503367
#> [2886] 39.174198 48.377148 339.486165 101.688007 149.401876
#> [2891] 84.371173 66.150104 48.432286 44.200528 48.981838
#> [2896] 350.977215 101.010195 155.124837 261.774278 62.327187
#> [2901] 50.881668 41.499536 58.946579 426.405576 137.709243
#> [2906] 154.974981 1044.061565 75.624113 52.398541 56.741179
#> [2911] 90.532370 475.939693 236.967228 139.114540 3432.023477
#> [2916] 110.743161 75.961875 92.011387 166.027562 929.845177
#> [2921] 504.843100 139.855029 6483.536127 197.809790 140.472021
#> [2926] 127.368047 224.242597 1451.459637 717.187880 141.928024
#> [2931] 7367.100911 239.956124 158.085961 124.970249 220.324988
#> [2936] 1630.745826 600.373785 130.757386 6646.782293 195.933957
#> [2941] 165.239449 116.567387 190.198294 1142.097216 483.831937
#> [2946] 129.864168 5204.742301 162.181947 194.907652 100.592671
#> [2951] 155.622761 852.478428 309.304721 137.013250 4042.596232
#> [2956] 117.024751 183.219997 75.718323 108.972406 623.185056
#> [2961] 276.796656 138.015189 2280.942842 83.652375 146.110538
#> [2966] 57.217777 85.910595 426.909449 169.369923 130.765452
#> [2971] 1129.015567 72.541058 98.417314 51.786954 65.490581
#> [2976] 370.740360 137.832735 128.707394 584.099025 70.200104
#> [2981] 78.216922 45.082211 53.395062 396.197642 115.825544
#> [2986] 164.890511 344.759542 67.780839 58.186826 49.007957
#> [2991] 57.555344 377.432551 103.528653 173.131749 304.128630
#> [2996] 82.324333 59.280267 49.017062 58.471389 385.087567
#> [3001] 107.713102 188.159390 256.629299 76.506054 144.285021
#> [3006] 51.495279 51.635400 381.894585 115.585556 176.218209
#> [3011] 207.990364 64.513575 471.014842 48.795036 46.370679
#> [3016] 413.799603 114.708741 170.739805 142.628879 51.914241
#> [3021] 766.677505 46.983835 45.447159 385.264893 105.968021
#> [3026] 157.320836 124.018762 52.331639 727.865570 45.855819
#> [3031] 48.050871 464.501195 108.529287 140.250347 114.867799
#> [3036] 49.210129 1365.406863 50.159966 48.346549 401.379074
#> [3041] 94.303394 127.682224 76.557818 38.802320 8245.265938
#> [3046] 75.312788 41.553529 443.388435 82.071571 97.633059
#> [3051] 55.690245 35.804840 11817.493575 99.572008 41.501168
#> [3056] 446.000312 72.960672 84.298512 48.043065 35.754181
#> [3061] 12414.736519 99.395533 38.658161 397.013831 74.672682
#> [3066] 72.898924 43.723220 44.488826 12981.508815 108.049080
#> [3071] 39.232533 376.557258 79.987398 79.204333 40.242510
#> [3076] 44.137902 13369.054391 87.335052 34.362953 431.964433
#> [3081] 80.404728 81.348712 39.098851 42.970071 12251.031919
#> [3086] 83.383473 40.176316 405.543253 73.572794 92.162209
#> [3091] 47.164285 47.335381 11181.342307 80.073486 33.482190
#> [3096] 381.244893 82.093875 106.339604 55.248650 41.232214
#> [3101] 10035.242946 67.145501 41.769664 351.287240 79.788459
#> [3106] 116.131377 54.850998 41.936423 7552.693637 64.331488
#> [3111] 45.096367 363.235391 88.494106 152.857911 58.406151
#> [3116] 45.085574 5274.329443 54.984343 51.276605 384.683981
#> [3121] 85.207682 170.325455 56.245315 48.942358 3446.959797
#> [3126] 62.191727 72.638760 376.092814 96.718939 207.073727
#> [3131] 51.516924 58.128504 2306.458394 105.223533 90.857087
#> [3136] 437.576096 91.756143 202.965217 50.084695 171.950652
#> [3141] 1713.140865 201.747017 165.403270 574.701225 97.888899
#> [3146] 224.355435 50.426250 643.298990 1559.863603 357.425180
#> [3151] 250.534715 1457.781432 77.966053 327.988844 48.599136
#> [3156] 1830.193673 2340.699961 361.455326 221.523901 3259.837324
#> [3161] 80.292646 667.704245 46.440136 3960.048515 3318.063234
#> [3166] 374.118420 251.656618 4836.139255 75.492559 911.828175
#> [3171] 45.894324 5028.931315 3790.007761 376.525573 204.266086
#> [3176] 6447.558959 81.661543 1083.513819 48.934917 5462.590695
#> [3181] 4040.078162 316.379302 201.778532 5451.933901 77.594644
#> [3186] 1029.314338 50.048455 4427.503115 4019.770812 354.163547
#> [3191] 159.297537 4770.406330 89.531190 681.997054 50.058454
#> [3196] 3735.746069 3014.155752 253.142876 138.573892 3442.385517
#> [3201] 108.520769 567.223315 48.193977 2389.961043 2112.600046
#> [3206] 167.179547 116.898048 2200.591211 90.379031 389.386558
#> [3211] 58.841952 1387.697525 1671.304308 122.009446 91.749983
#> [3216] 1334.821787 86.827529 273.706301 47.739604 682.037909
#> [3221] 2702.578604 116.541629 68.538327 1053.298147 90.165921
#> [3226] 199.887662 48.969506 366.617997 8110.564172 119.166472
#> [3231] 57.492952 1253.169747 79.696759 125.325973 70.386734
#> [3236] 185.103845 17817.324496 154.758688 54.444984 1848.090470
#> [3241] 86.306052 112.925436 69.415037 97.107928 24421.982208
#> [3246] 177.387707 53.053684 2350.667822 68.231133 115.283247
#> [3251] 70.470103 67.408725 24224.176889 199.367430 54.483140
#> [3256] 1851.521975 71.252109 104.026548 73.399323 62.840590
#> [3261] 20173.169426 159.624412 54.335656 1643.522781 75.692504
#> [3266] 112.373523 84.076986 71.224743 15413.772897 94.253645
#> [3271] 46.128246 1353.106928 87.476977 123.569037 76.496362
#> [3276] 83.747586 10410.026459 73.003667 43.600309 914.755517
#> [3281] 86.775649 147.780305 77.343147 80.636035 5586.789067
#> [3286] 59.589081 41.941584 542.935288 94.678388 156.837136
#> [3291] 111.355258 84.365073 2892.251637 49.823283 51.304918
#> [3296] 529.727005 106.611109 202.748799 118.293521 114.514784
#> [3301] 1277.830641 57.576321 46.114399 422.564773 105.676005
#> [3306] 216.152271 131.173524 98.990215 730.104704 56.522926
#> [3311] 53.390978 384.438329 93.755092 285.822187 97.594344
#> [3316] 85.205392 547.206071 46.915710 51.918508 404.797401
#> [3321] 93.750448 295.711001 140.678887 77.257286 421.634536
#> [3326] 46.707930 54.991718 377.567921 100.374922 312.936990
#> [3331] 132.502633 71.580301 274.483526 49.194447 61.477936
#> [3336] 431.160271 88.113807 354.666093 124.343121 70.724463
#> [3341] 237.235026 53.864975 57.278502 386.820263 99.928820
#> [3346] 387.976732 135.955634 88.630018 214.309572 54.529801
#> [3351] 54.718577 337.031514 114.828256 287.822821 156.152333
#> [3356] 119.974355 180.442025 69.066942 51.631361 386.171758
#> [3361] 110.462710 307.941976 156.382602 142.630032 144.549083
#> [3366] 53.404418 50.098636 364.021570 140.529983 246.337782
#> [3371] 215.574043 150.186601 143.793425 44.319647 49.858377
#> [3376] 425.172901 146.985848 220.863097 398.886785 190.947449
#> [3381] 121.991410 53.573488 47.726592 383.298964 129.257191
#> [3386] 217.825355 747.478983 243.704667 125.857035 54.983686
#> [3391] 50.734335 448.644560 128.739227 227.458562 1514.914099
#> [3396] 446.209846 125.520585 50.398286 59.134788 446.003391
#> [3401] 107.173052 231.371720 2620.309723 705.822113 115.209906
#> [3406] 53.223066 70.486035 459.659304 106.569502 337.338604
#> [3411] 4899.363778 1153.960002 134.494480 56.529112 78.164909
#> [3416] 583.802260 104.608224 499.497467 7395.244639 1887.052951
#> [3421] 211.215561 94.945173 103.651928 788.532002 106.056672
#> [3426] 800.572374 10043.866250 2623.693823 240.230819 111.280702
#> [3431] 148.987083 914.847067 109.614644 1249.143255 12097.695780
#> [3436] 3653.450436 277.743610 182.055874 179.234241 1221.271458
#> [3441] 126.401723 1715.142546 14563.587860 5407.566131 264.512774
#> [3446] 210.784998 229.734521 1587.126612 124.967387 2232.878777
#> [3451] 15532.657079 6234.608575 269.324034 208.424586 233.263276
#> [3456] 1552.763248 120.786471 1960.332508 15743.664269 5880.225753
#> [3461] 220.480492 230.471743 231.182513 1374.267268 122.930560
#> [3466] 1785.462605 13040.903736 4625.013483 185.892917 177.027535
#> [3471] 207.044462 1064.031657 129.958451 1151.740920 10275.138221
#> [3476] 3030.566003 134.904623 101.688033 129.392091 737.169609
#> [3481] 110.267769 682.744596 7212.835770 1644.275912 96.058334
#> [3486] 74.493444 88.870497 520.998709 98.802609 438.159170
#> [3491] 3896.246661 903.259424 80.395037 70.742299 64.676344
#> [3496] 463.621414 92.772626 298.435963 2059.432492 466.110953
#> [3501] 74.424627 65.230299 55.563819 424.500233 95.895749
#> [3506] 218.235428 1111.334132 276.446303 70.360008 58.971164
#> [3511] 51.898553 421.426599 94.400382 183.128169 641.387997
#> [3516] 186.855560 64.828225 61.017041 46.322119 405.869448
#> [3521] 96.276210 165.488380 444.988146 134.354429 68.978034
#> [3526] 59.648629 41.928908 390.248778 97.680430 142.669647
#> [3531] 326.401134 100.406484 69.730199 56.872277 47.295864
#> [3536] 410.286925 100.654992 147.297034 253.873683 88.259557
#> [3541] 69.272909 54.819753 53.691147 357.119705 96.424177
#> [3546] 159.983678 197.203818 70.524063 64.887608 59.124792
#> [3551] 55.327900 392.013416 112.728092 166.700046 165.703903
#> [3556] 70.800969 71.877032 53.680604 150.170933 386.079010
#> [3561] 128.308721 142.192752 116.723960 63.349980 94.577026
#> [3566] 52.785717 618.750584 350.651580 115.429550 162.722968
#> [3571] 86.215457 51.653908 203.842317 53.626194 2246.121891
#> [3576] 382.415158 102.911678 134.274657 65.262824 67.883797
#> [3581] 529.226182 68.566852 6140.326677 470.275514 86.503963
#> [3586] 120.076985 52.290525 95.471138 1327.676572 152.403422
#> [3591] 12409.546012 446.365777 81.111320 120.051437 38.316607
#> [3596] 125.396044 2111.053553 192.086990 15265.605845 505.665088
#> [3601] 68.267354 110.300507 42.960764 122.990922 2434.511379
#> [3606] 193.448085 13856.641640 473.746327 61.488787 118.927335
#> [3611] 40.846413 98.481253 2635.015115 145.921496 10782.008108
#> [3616] 514.134987 75.622974 108.849099 43.482325 72.364911
#> [3621] 3990.963925 135.316360 7795.630482 619.633191 76.814228
#> [3626] 96.248114 42.018788 61.545582 5406.427674 89.845524
#> [3631] 4769.421076 970.879131 75.477365 85.024211 40.636458
#> [3636] 56.770813 5599.366446 84.311730 2351.550069 1181.581148
#> [3641] 78.535752 87.937028 51.470706 80.762274 5635.438907
#> [3646] 160.984848 1184.728514 1395.521664 100.176642 86.781772
#> [3651] 73.051767 160.614398 5912.221951 219.380200 738.510947
#> [3656] 1140.732565 145.020645 84.483414 86.219270 289.808744
#> [3661] 4809.441537 317.619402 519.229572 955.273766 138.266457
#> [3666] 86.975240 94.872176 315.805714 4244.719148 287.564623
#> [3671] 385.001572 745.416471 139.109839 94.263892 89.964926
#> [3676] 245.282922 3344.973796 379.042695 285.002069 562.866452
#> [3681] 118.547819 93.237187 90.176100 196.391278 2100.318811
#> [3686] 372.299956 239.873667 524.349564 106.931143 106.563291
#> [3691] 76.841937 139.732984 1207.312222 316.286884 197.681512
#> [3696] 436.388388 91.078274 113.614040 71.921476 96.063303
#> [3701] 652.662973 256.810664 168.067726 441.319650 88.802723
#> [3706] 109.248382 81.351967 73.164431 396.452972 225.406458
#> [3711] 142.344921 374.552271 93.302201 111.343348 67.648089
#> [3716] 70.830980 300.911453 204.240726 133.175767 342.959788
#> [3721] 87.580712 113.913341 69.461376 65.748910 411.875001
#> [3726] 308.109618 129.885858 418.053823 88.435850 126.543285
#> [3731] 58.067230 82.053081 1163.347622 656.206055 142.131827
#> [3736] 483.019800 81.782910 125.925370 55.778440 134.806233
#> [3741] 4152.926938 1369.659515 175.497921 1422.080153 74.546009
#> [3746] 142.809977 57.799691 158.359331 10781.055266 1887.565770
#> [3751] 254.454628 4125.279513 57.171463 159.062548 49.352282
#> [3756] 191.586048 19414.716845 1915.301956 249.685607 9593.457980
#> [3761] 53.229879 179.321117 49.398018 219.538918 25027.612744
#> [3766] 1699.348942 237.978199 12125.899330 47.666146 150.118401
#> [3771] 45.483353 205.384385 24722.054114 1560.084209 187.063079
#> [3776] 10025.053218 47.770314 142.855425 45.881075 160.115075
#> [3781] 22247.285425 1122.209056 179.858794 7811.386646 57.296257
#> [3786] 139.605985 42.877661 120.245360 17227.041343 966.745916
#> [3791] 160.845884 4808.898843 69.792791 125.729821 38.167593
#> [3796] 84.820290 11515.139382 548.766364 115.779634 2597.306317
#> [3801] 74.623623 115.060676 45.678531 75.170218 7086.094049
#> [3806] 378.804598 96.321522 1232.934934 81.323214 120.188202
#> [3811] 56.723953 66.465334 3185.372583 268.484301 82.963581
#> [3816] 687.802587 85.506730 117.224576 64.479412 65.524019
#> [3821] 1814.608266 225.544707 85.407348 542.782638 86.349111
#> [3826] 123.004558 58.317664 66.202576 1100.415739 214.043717
#> [3831] 77.217544 388.616710 81.948864 124.003550 70.615695
#> [3836] 61.841084 667.588209 172.941852 72.987441 404.906494
#> [3841] 91.581221 116.061535 69.550928 61.093073 432.550454
#> [3846] 190.066188 68.221521 423.191811 86.533175 132.812863
#> [3851] 63.531707 67.118890 403.301821 192.687840 73.299575
#> [3856] 444.097988 89.506922 113.664797 60.807217 65.998789
#> [3861] 407.663872 164.538277 124.435118 553.638583 86.238665
#> [3866] 130.324484 70.253929 61.002073 435.633906 152.830988
#> [3871] 391.973385 547.802014 81.861402 112.163967 59.381364
#> [3876] 59.971323 411.557948 135.541161 1744.958889 612.703115
#> [3881] 88.034656 110.804463 61.708663 71.230467 293.507325
#> [3886] 110.757471 6077.740919 862.483115 81.360095 105.068825
#> [3891] 67.573796 81.850953 186.911564 111.805663 13307.111044
#> [3896] 1372.664704 70.698866 88.051398 82.856088 108.482019
#> [3901] 144.194354 85.800517 18869.675912 1635.211647 64.893472
#> [3906] 88.788734 104.189162 113.902982 109.419341 84.412407
#> [3911] 20187.201067 1944.522540 63.006302 102.301590 93.458797
#> [3916] 111.017181 99.386746 89.749888 18506.676232 1730.000063
#> [3921] 62.605072 139.625281 84.196161 92.953162 83.759750
#> [3926] 76.876001 14237.193458 1203.023799 84.155309 189.537974
#> [3931] 77.877443 70.071738 80.401970 103.007223 8360.494762
#> [3936] 828.841696 84.691190 241.267317 65.904531 64.646327
#> [3941] 83.996235 113.440815 4721.723304 635.543387 90.308433
#> [3946] 222.556461 53.405444 55.582060 78.115123 125.319860
#> [3951] 2118.763134 487.729132 93.171913 192.335782 61.750752
#> [3956] 56.489347 108.363090 132.220562 932.305071 423.365816
#> [3961] 91.165989 145.139294 80.454267 51.459135 306.211543
#> [3966] 72.197537 439.134534 393.756957 76.900078 172.189370
#> [3971] 112.221617 34.701941 937.758675 87.240965 236.845782
#> [3976] 579.096423 102.674654 293.163169 327.418643 34.228399
#> [3981] 3036.340119 137.194945 179.647357 1120.911737 109.453593
#> [3986] 971.508357 1178.908284 39.262406 7097.736498 177.924146
#> [3991] 192.667422 2053.846316 278.958163 1660.497186 1904.867305
#> [3996] 47.415967 13612.566461 342.179352 294.165621 3160.448690
#> [4001] 435.425394 2127.446369 2887.688182 42.565342 14766.058057
#> [4006] 345.222552 278.494640 3322.225743 372.767125 2362.250764
#> [4011] 2609.591924 38.852476 12625.212351 309.212933 222.647444
#> [4016] 2664.950264 315.022733 1834.223903 1730.079802 38.026996
#> [4021] 9392.282426 224.407183 190.014574 1941.780229 207.846138
#> [4026] 1009.193277 1040.100975 40.215923 5706.949405 163.142562
#> [4031] 250.473155 1476.288686 148.233481 633.366188 545.469896
#> [4036] 40.898872 3206.350817 153.621670 1082.551877 1200.967449
#> [4041] 119.793940 425.444598 296.827609 42.566649 1538.463505
#> [4046] 268.504453 3411.768063 1179.528350 117.978419 465.694062
#> [4051] 182.231784 59.051107 707.841693 798.219025 5707.023102
#> [4056] 1486.346857 117.639523 831.651246 192.140575 69.683965
#> [4061] 487.604232 1810.516731 5655.658358 1759.984767 138.435596
#> [4066] 908.943295 205.512557 62.792249 586.376364 3350.792698
#> [4071] 3557.345636 2794.186745 96.937053 1355.829542 229.861769
#> [4076] 122.015590 1829.677277 6027.546728 3410.287913 2622.834168
#> [4081] 77.087682 1324.298012 191.503052 163.245090 3664.284597
#> [4086] 5437.512162 1735.487584 2429.998068 108.438155 1178.696880
#> [4091] 162.410241 267.114850 6283.259947 4333.732537 1024.766212
#> [4096] 1805.691424 108.535899 706.189219 91.852790 244.940871
#> [4101] 8028.558449 2034.124100 643.886912 1045.987358 68.133528
#> [4106] 330.332173 52.346737 154.506984 8406.522748 1084.783013
#> [4111] 396.354323 536.963676 54.900689 156.037455 36.911622
#> [4116] 100.574239 8086.313752 465.948604 289.557508 323.240735
#> [4121] 51.519895 121.549670 37.724636 70.075937 7054.943526
#> [4126] 284.338271 164.873854 264.384663 44.183074 94.368913
#> [4131] 33.475381 53.233161 5233.526518 206.557768 148.569036
#> [4136] 229.047964 47.795181 101.349570 35.298529 43.934168
#> [4141] 3946.345265 164.245997 129.941261 229.565143 52.575088
#> [4146] 81.682126 40.134699 42.011162 2149.719056 180.495567
#> [4151] 153.656255 252.291856 51.914673 96.644777 39.215569
#> [4156] 44.699284 1105.301500 170.612759 191.196159 299.601564
#> [4161] 55.762865 89.791462 37.016342 48.470076 631.509387
#> [4166] 142.981072 313.874315 261.312555 54.545496 96.572753
#> [4171] 38.547161 40.580551 403.145616 144.833464 342.246587
#> [4176] 310.954171 64.032320 98.969568 41.323106 47.257789
#> [4181] 320.515738 136.787292 286.709227 266.342598 53.949720
#> [4186] 100.135118 41.356455 43.328885 267.502695 144.459614
#> [4191] 164.988137 328.513179 57.461130 94.882088 40.005957
#> [4196] 39.564587 207.814849 126.104782 119.881971 280.057880
#> [4201] 52.793556 89.049451 35.078434 46.238785 146.926702
#> [4206] 110.740945 84.288822 279.134654 47.290907 94.405094
#> [4211] 37.226860 48.481465 162.433349 106.836158 77.830367
#> [4216] 264.453547 64.449780 88.148473 51.289629 40.248770
#> [4221] 127.812709 104.602114 67.768942 258.024133 61.969323
#> [4226] 82.904072 41.338179 42.952148 110.149472 117.332622
#> [4231] 67.180968 204.606299 56.525766 90.216799 46.333286
#> [4236] 44.435855 99.924897 100.880076 64.386607 258.448507
#> [4241] 66.325437 93.075675 36.416222 40.134242 95.424911
#> [4246] 120.528329 62.859443 261.412888 61.412603 86.133227
#> [4251] 42.906540 39.572776 82.769467 111.134468 51.519102
#> [4256] 286.668917 64.283166 84.813246 40.286968 37.904904
#> [4261] 69.903974 101.585087 56.501014 278.245946 61.309376
#> [4266] 87.758078 39.301138 41.492513 65.751722 104.708166
#> [4271] 52.745788 289.672285 62.170847 84.853457 36.613074
#> [4276] 41.065433 63.258503 105.844881 55.619390 279.935325
#> [4281] 65.660522 80.181659 38.893459 40.557632 57.631244
#> [4286] 128.399536 52.285071 293.503915 56.622564 95.275258
#> [4291] 45.338733 45.787694 54.856984 98.758217 53.656562
#> [4296] 293.183126 46.206202 110.215465 74.328091 56.813193
#> [4301] 66.842069 118.049178 58.332928 357.771687 56.772910
#> [4306] 123.815829 89.686368 71.647151 56.830797 153.497084
#> [4311] 49.366267 331.818114 56.743310 116.216086 76.397035
#> [4316] 59.559612 59.977540 145.390264 49.855219 314.519660
#> [4321] 61.537938 121.968848 67.554024 61.811850 57.497588
#> [4326] 129.414189 49.220161 280.976827 58.198151 109.382159
#> [4331] 58.828400 63.334645 51.710396 158.309001 53.081195
#> [4336] 233.696873 54.915254 99.290802 37.056606 53.601216
#> [4341] 50.327137 161.450047 65.423462 192.412946 50.879132
#> [4346] 92.203203 51.482459 50.083648 51.578465 137.690177
#> [4351] 82.735760 282.085141 49.772270 92.265072 68.277279
#> [4356] 73.567708 58.589771 142.897967 86.865603 321.770474
#> [4361] 56.413381 206.576746 171.211971 131.399072 46.079827
#> [4366] 159.835466 177.649695 363.398232 56.437854 584.305911
#> [4371] 571.609175 323.764918 46.295060 228.783927 426.355290
#> [4376] 542.757191 46.243221 1658.880072 1622.507155 997.029544
#> [4381] 52.807188 531.293593 749.134423 922.991166 39.452485
#> [4386] 2825.488790 2877.784777 1508.763285 65.139674 831.675763
#> [4391] 1141.031608 1391.305612 39.270631 3856.434958 3860.638527
#> [4396] 2222.188867 71.057165 1332.707846 1264.313461 1480.532561
#> [4401] 40.535523 3580.879003 3477.841466 2148.575595 77.281534
#> [4406] 1014.496104 1115.037402 1125.428625 39.748277 2954.830371
#> [4411] 2742.905155 1478.568493 61.673042 600.030137 762.467306
#> [4416] 751.502703 42.944433 2058.174511 1747.792682 818.145429
#> [4421] 47.708205 379.688054 512.696804 591.761472 42.598034
#> [4426] 1085.150648 920.500510 370.857232 43.804657 257.972650
#> [4431] 373.951200 475.887365 55.008088 646.133952 497.478089
#> [4436] 222.843946 41.230330 194.674755 178.710725 371.505328
#> [4441] 57.140406 339.753634 231.120987 134.328742 42.509141
#> [4446] 153.446260 132.452704 337.295803 54.562566 238.367276
#> [4451] 181.902731 99.626090 45.039130 152.466819 123.765795
#> [4456] 319.224321 53.479184 191.147905 133.862770 77.500712
#> [4461] 40.884697 144.562822 89.399850 310.357274 50.228462
#> [4466] 148.823911 97.549715 72.132407 39.522509 130.002939
#> [4471] 77.423112 289.631985 55.407644 142.645613 103.648934
#> [4476] 71.219004 51.694390 139.958523 82.921971 292.494079
#> [4481] 52.205973 181.637192 69.654248 81.094329 68.157062
#> [4486] 169.031478 86.042279 308.772410 53.377927 310.139904
#> [4491] 69.020225 66.398735 76.538431 163.269922 74.792746
#> [4496] 264.083464 48.114779 829.376209 69.830268 66.752727
#> [4501] 76.916664 163.950370 86.172492 276.670364 52.656940
#> [4506] 1914.579471 65.068199 58.827195 81.856336 196.329029
#> [4511] 104.180491 502.488765 53.568452 2747.098598 51.924475
#> [4516] 59.908836 87.904676 202.476039 125.925033 1019.848396
#> [4521] 53.380118 3511.184929 48.108025 56.457501 150.473284
#> [4526] 227.412918 142.088304 2872.239379 56.085864 3064.459565
#> [4531] 70.128100 63.608724 241.978705 214.168285 127.073370
#> [4536] 7327.306561 47.353124 2551.168664 69.940965 78.336143
#> [4541] 241.413151 249.864527 163.291762 11816.092896 44.269472
#> [4546] 1506.343634 55.429840 75.701900 178.393479 213.936204
#> [4551] 149.746016 10828.569943 51.917788 875.938513 50.516516
#> [4556] 91.879225 128.072299 243.998080 124.920143 8187.996344
#> [4561] 52.379129 452.274888 42.016104 67.711327 86.443328
#> [4566] 215.559648 101.002137 4219.553405 51.288061 276.236507
#> [4571] 38.036253 62.686607 70.009414 187.261870 91.053534
#> [4576] 1847.232543 55.696516 199.953871 36.243559 58.281916
#> [4581] 57.880514 191.821269 82.974095 941.544817 50.940288
#> [4586] 163.811216 38.687191 51.246496 49.906409 169.517866
#> [4591] 73.059557 643.602890 55.092397 133.211018 39.873766
#> [4596] 49.214188 48.038795 168.921205 67.181868 493.367367
#> [4601] 50.925881 118.515011 31.855963 52.775020 46.840387
#> [4606] 179.739927 69.377023 411.059316 50.439892 112.860340
#> [4611] 32.885993 50.016701 48.796571 166.045830 63.744490
#> [4616] 379.992069 49.849600 125.535197 36.394381 42.845141
#> [4621] 44.649761 183.210385 67.217464 317.219016 55.596707
#> [4626] 92.889999 34.362905 50.603551 48.219711 200.643150
#> [4631] 58.318596 312.149223 49.672869 92.801931 31.449582
#> [4636] 48.793051 45.627728 183.778058 52.802457 302.197141
#> [4641] 51.193953 80.921134 39.615357 46.794926 47.705001
#> [4646] 180.875624 52.438282 276.114152 50.887574 82.552679
#> [4651] 36.917704 47.417947 40.085574 175.438082 61.262524
#> [4656] 286.949399 53.042678 103.432709 33.243855 53.685708
#> [4661] 49.722538 185.507442 65.283741 287.849976 52.636642
#> [4666] 84.107162 31.474252 63.818638 48.940789 164.824659
#> [4671] 70.795252 264.853286 59.222573 90.152780 27.160904
#> [4676] 89.749870 57.063146 181.639975 66.890297 269.435757
#> [4681] 66.396097 80.147617 28.577496 122.774279 71.885516
#> [4686] 178.217865 59.325944 247.322788 68.980612 85.526426
#> [4691] 30.771644 111.374158 85.778464 230.254863 59.405668
#> [4696] 262.859357 56.453945 80.841921 25.095487 111.384500
#> [4701] 101.615747 292.653718 75.714277 236.219201 62.770011
#> [4706] 76.347353 29.549898 92.813874 94.254533 277.517573
#> [4711] 53.902052 232.997644 59.397059 71.790755 29.417685
#> [4716] 80.426822 88.550111 244.533126 56.060521 237.362021
#> [4721] 55.113275 72.179748 30.059435 68.114589 64.858636
#> [4726] 246.275348 48.147082 231.276261 59.358420 71.755033
#> [4731] 31.014290 61.453193 56.600685 207.804537 44.582336
#> [4736] 216.942810 58.470191 74.902481 29.037620 56.128376
#> [4741] 41.433763 214.985116 47.945246 238.681643 54.878736
#> [4746] 74.306264 28.871119 53.933283 51.477292 240.338017
#> [4751] 45.071880 319.892479 60.243923 71.908718 30.792017
#> [4756] 50.461645 43.276440 228.400380 49.487292 296.458126
#> [4761] 46.205151 72.394365 31.631879 51.444877 44.081211
#> [4766] 264.518612 43.777099 331.151428 52.130969 75.112764
#> [4771] 34.393221 46.578332 42.388637 276.102014 56.115048
#> [4776] 362.837851 58.398589 74.458122 30.720373 45.265009
#> [4781] 44.877775 276.971183 52.895040 349.033458 46.847567
#> [4786] 72.428151 34.847921 63.150338 52.859219 278.265463
#> [4791] 53.059548 396.797879 47.275672 66.330887 29.480862
#> [4796] 71.637194 53.837807 307.917395 56.975557 497.815422
#> [4801] 48.149000 72.074457 30.924934 93.585150 44.845495
#> [4806] 333.082104 64.602899 628.595244 46.241044 68.278919
#> [4811] 40.911953 91.214876 49.440920 293.720754 60.020386
#> [4816] 627.625701 50.614990 69.169435 28.467986 70.115238
#> [4821] 43.505653 309.938003 59.928173 575.279742 47.659137
#> [4826] 71.464793 32.106199 60.286711 49.790274 294.248272
#> [4831] 58.653218 685.388791 46.213836 69.006738 29.242110
#> [4836] 51.410295 45.520157 357.318091 51.821616 703.832977
#> [4841] 47.358509 75.602755 28.371720 51.350037 48.103827
#> [4846] 420.375025 54.557280 660.463500 55.045969 76.902699
#> [4851] 32.517357 51.046595 47.326629 455.927849 46.463520
#> [4856] 652.636737 49.820302 67.005941 32.962791 56.354765
#> [4861] 39.792577 535.631324 50.786102 482.542554 45.937160
#> [4866] 66.954484 27.526334 57.859302 47.467405 676.631754
#> [4871] 47.187554 475.886387 45.488405 68.867610 28.043411
#> [4876] 66.464158 53.735365 990.586320 48.262246 486.732631
#> [4881] 47.613015 64.115459 26.575104 89.239618 78.133335
#> [4886] 2147.019938 60.448976 485.854286 46.575728 66.852067
#> [4891] 25.055291 140.143325 119.297522 4785.156591 79.674124
#> [4896] 468.269479 40.069633 53.752282 21.424326 230.675430
#> [4901] 179.378576 6983.051321 108.451029 604.748155 36.021672
#> [4906] 56.476180 25.989365 277.886080 238.432355 8121.600158
#> [4911] 113.564386 529.230850 34.702434 56.259035 24.925178
#> [4916] 263.869149 208.314547 6791.957569 115.353383 462.317766
#> [4921] 42.774459 62.295376 25.392242 182.715289 113.173815
#> [4926] 4407.928285 90.005843 380.251795 46.900266 55.914632
#> [4931] 27.799285 115.570539 79.058044 2517.681085 67.723625
#> [4936] 365.169886 46.346011 63.736670 26.285887 63.575800
#> [4941] 54.973395 1016.756863 59.035719 314.809195 42.061985
#> [4946] 57.064270 24.355705 49.483228 41.642904 629.549182
#> [4951] 46.952355 299.548207 43.353903 61.252663 28.244145
#> [4956] 44.116027 40.359105 413.014250 42.694832 348.094255
#> [4961] 44.406247 60.548911 23.920501 41.520794 37.626969
#> [4966] 280.745009 44.364923 276.202102 49.087521 69.952603
#> [4971] 29.107688 37.645823 37.956093 187.936462 49.330026
#> [4976] 271.683895 49.996027 72.130079 24.524474 36.834286
#> [4981] 34.680823 173.507615 48.710235 308.370965 49.999272
#> [4986] 65.250505 27.344688 37.068941 29.006739 126.356819
#> [4991] 53.180806 325.906607 45.791061 65.085692 30.906553
#> [4996] 42.035999 33.820820 115.196643 48.461630 282.270670
#> [5001] 47.862877 65.823179 27.003224 36.460184 31.905152
#> [5006] 86.673064 53.963977 334.402929 45.000086 68.865902
#> [5011] 26.074596 33.596923 34.060453 76.903663 56.431894
#> [5016] 328.876977 45.333796 61.137680 25.360921 34.779504
#> [5021] 35.907375 71.257939 54.631313 321.782398 55.947935
#> [5026] 75.419724 26.057713 33.953019 33.511435 62.095741
#> [5031] 48.594550 318.331998 50.517598 62.658040 26.447861
#> [5036] 35.184809 35.683931 49.447750 45.818880 297.853359
#> [5041] 43.528390 64.768476 25.413464 30.008566 33.274146
#> [5046] 54.379836 43.844371 277.684504 47.210207 61.803186
#> [5051] 28.459141 36.008184 32.183616 45.571559 43.043387
#> [5056] 216.424570 47.089564 61.269947 26.100267 31.252960
#> [5061] 30.096715 43.530337 63.426359 240.832936 45.444751
#> [5066] 69.823334 23.452219 34.694233 28.758334 43.586328
#> [5071] 228.805007 271.242768 51.764197 59.215860 25.704444
#> [5076] 35.203498 33.849074 57.042047 883.215340 198.589383
#> [5081] 46.525925 59.050193 25.690238 33.508681 34.143498
#> [5086] 103.191358 3793.034272 237.970979 46.374508 63.158940
#> [5091] 23.593530 34.508682 37.986472 321.577456 10387.647943
#> [5096] 221.502027 46.100283 40.960708 20.854672 40.466064
#> [5101] 48.014631 458.881079 14625.467060 334.037362 33.822183
#> [5106] 49.832747 22.347413 43.504691 51.173487 664.266095
#> [5111] 16593.904326 282.950405 31.236959 43.570580 20.728093
#> [5116] 43.185005 59.566814 676.587390 16188.642222 313.413631
#> [5121] 38.075133 51.941585 14.116758 49.725098 60.537468
#> [5126] 490.664727 12504.341366 332.336475 37.057482 50.585754
#> [5131] 25.794391 34.584245 39.173538 294.094119 9368.650824
#> [5136] 238.538071 39.816894 53.761216 27.356094 42.350012
#> [5141] 32.642037 161.953584 4959.686799 273.161323 41.500725
#> [5146] 72.503669 22.130488 33.884160 28.541240 80.038212
#> [5151] 1901.186338 188.447495 53.187130 69.078084 33.401141
#> [5156] 33.780515 33.022667 61.955306 1182.946692 215.098218
#> [5161] 42.515117 62.778956 26.216766 35.221615 48.032176
#> [5166] 54.959703 825.698900 207.598380 55.643398 71.384935
#> [5171] 31.635770 35.349741 48.964816 50.205919 578.695492
#> [5176] 265.714340 42.320107 61.631955 24.768630 33.323584
#> [5181] 48.746423 39.307374 432.141679 328.384663 49.987269
#> [5186] 60.807526 28.164686 37.712182 35.494426 33.273868
#> [5191] 327.161537 237.954949 51.004126 62.141679 24.341466
#> [5196] 33.163396 32.038287 57.151888 307.955087 293.975123
#> [5201] 45.686545 60.149132 22.322665 32.619832 39.601874
#> [5206] 23.049419 240.837605 186.115206 46.597708 51.809865
#> [5211] 30.635418 21.868757 36.862236 31.747517 177.808305
#> [5216] 258.993183 35.778968 80.982385 17.748673 31.080975
#> [5221] 27.910608 29.008353 152.458723 206.890375 60.902021
#> [5226] 55.154165 30.905965 35.537998 26.481802 36.547885
#> [5231] 71.281653 319.363948 39.509593 54.674671 23.694966
#> [5236] 22.768565 35.902006 15.519170 122.210194 311.873757
#> [5241] 54.575711 52.414871 43.260145 27.726636 26.703534
#> [5246] 29.005692 49.958104 318.460082 32.422931 65.452027
#> [5251] 16.243843 29.931222 67.388561 21.198202 115.150943
#> [5256] 311.011074 49.097036 34.894409 32.663204 26.605071
#> [5261] 143.219070 132.124666 52.465763 726.977373 24.659910
#> [5266] 50.157988 9.839966 34.117595 213.281965 367.988658
#> [5271] 55.975199 1133.979505 35.030130 48.212218 18.222072
#> [5276] 27.248220 749.983686 568.723259 73.905793 1688.907677
#> [5281] 28.385296 40.115522 9.298693 25.190910 284.538703
#> [5286] 1371.456125 51.937697 1970.014395 31.139139 30.812823
#> [5291] 17.957888 20.077910 2250.042225 247.380044 120.841294
#> [5296] 358.835250 29.951044 30.655786 10.936186 22.041673
#> [5301] 842.268770 684.991122 50.578106 751.022704 17.030382
#> [5306] 56.704069 9.993893 26.125907 395.690440 108.014778
#> [5311] 52.044293 173.997155 27.761691 56.691786 8.355937
#> [5316] 25.420549 121.274068 109.111402 40.842086 185.723421
#> [5321] 19.028397 54.993547 8.221156 30.516583 74.598682
#> [5326] 54.938835 47.603433 208.145478 26.427559 48.410746
#> [5331] 11.010037 29.746014 95.137569 31.070524 62.041933
#> [5336] 81.034690 27.960036 50.010338 13.210586 25.622529
#> [5341] 62.798812 34.743141 57.948189 67.948480 29.345248
#> [5346] 43.847262 10.212893 24.417106 37.715088 24.016516
#> [5351] 51.844956 56.948543 23.039180 47.985173 15.485633
#> [5356] 17.196868 28.604007 28.672823 49.094864 85.515405
#> [5361] 20.712237 54.197844 9.996985 24.535078 30.869278
#> [5366] 33.406760 37.092492 108.928467 29.427018 40.057003
#> [5371] 11.233572 25.663400 26.074822 14.684448 44.153481
#> [5376] 79.863051 20.674375 53.945479 8.635538 21.866742
#> [5381] 21.568681 27.295084 37.782676 119.027245 19.828372
#> [5386] 64.745642 6.866438 37.820110 16.976812 31.202158
#> [5391] 28.064303 126.817124 18.990707 62.140496 7.116460
#> [5396] 29.441396 13.949700 37.203920 24.919200 207.062197
#> [5401] 29.096854 47.315740 14.389914 27.333264 21.161312
#> [5406] 22.028162 24.699609 255.689930 37.234862 35.792454
#> [5411] 19.542832 31.829631 31.247314 19.193122 39.076528
#> [5416] 151.820509 35.495920 47.397914 20.446395 31.425314
#> [5421] 34.012810 15.253335 31.759759 121.802695 31.030318
#> [5426] 45.767372 12.877493 24.125255 27.306708 18.044403
#> [5431] 30.132601 109.521549 41.443886 49.541559 14.060000
#> [5436] 19.674713 30.753903 13.244524 32.146125 147.121849
#> [5441] 37.313982 45.717337 20.340095 23.995267 22.475538
#> [5446] 15.280520 30.612373 126.313107 34.105550 45.470899
#> [5451] 13.900067 23.654804 24.500477 16.061011 30.066356
#> [5456] 144.995349 35.720150 52.332116 22.641242 21.791053
#> [5461] 23.008396 17.488522 26.974505 117.847114 30.733337
#> [5466] 40.035589 13.306863 24.808838 22.640671 11.095066
#> [5471] 37.035494 169.536042 34.706612 42.308956 11.265198
#> [5476] 25.540988 17.534398 13.917723 36.879266 95.253177
#> [5481] 28.148921 54.359477 12.156747 23.301489 18.149539
#> [5486] 9.520780 25.295166 108.913805 31.419713 34.356447
#> [5491] 13.770139 23.333087 18.118756 9.574440 29.141270
#> [5496] 95.353999 35.310113 34.061251 13.283332 22.279662
#> [5501] 17.767520 10.718367 28.965177 93.648825 31.672368
#> [5506] 50.220073 13.240031 19.169652 16.961605 10.676379
#> [5511] 27.901531 101.624876 30.922475 30.711283 10.036392
#> [5516] 23.545922 15.679841 23.386392 32.812637 88.778950
#> [5521] 32.032750 38.288512 11.881548 17.974256 16.239587
#> [5526] 11.168106 24.564803 100.224677 27.833303 57.880141
#> [5531] 9.091872 15.109380 14.008010 10.726497 35.164280
#> [5536] 97.430139 27.099293 37.656634 11.423134 19.898495
#> [5541] 19.736534 10.571789 29.370398 124.398256 27.596925
#> [5546] 33.851647 11.234983 16.486515 15.795668 13.592158
#> [5551] 27.078752 93.896182 31.460622 44.212066 9.682059
#> [5556] 19.535536 14.092844 9.353039 32.686904 79.027967
#> [5561] 28.658147 38.346754 12.294601 14.046168 16.102160
#> [5566] 10.279072 34.637368 52.164251 28.343455 34.719642
#> [5571] 12.085510 16.228113 15.652985 9.625466 32.608459
#> [5576] 50.489848 31.156872 33.528733 10.087299 16.134050
#> [5581] 13.856099 7.972964 36.169426 88.756489 28.420947
#> [5586] 34.800597 9.344251 14.378970 15.781564 6.902752
#> [5591] 31.046286 59.280232 22.718516 32.093968 7.078729
#> [5596] 15.966721 14.978711 8.551211 28.791502 61.168031
#> [5601] 28.751535 34.515249 10.039587 15.328628 13.471790
#> [5606] 11.967418 22.615952 59.452777 25.812509 34.832149
#> [5611] 14.232937 18.010681 13.874654 12.665227 25.134650
#> [5616] 81.158182 25.747741 36.183789 9.222818 13.347133
#> [5621] 18.191310 5.800864 23.016008 59.047435 23.624210
#> [5626] 36.489993 8.455181 13.820350 20.218870 14.415679
#> [5631] 21.961414 42.435816 22.019617 24.294659 8.502477
#> [5636] 14.249668 21.836311 11.240619 31.662255 53.946034
#> [5641] 43.582236 29.396582 10.156057 13.793700 14.460816
#> [5646] 6.621688 23.156595 41.370287 23.171764 39.400401
#> [5651] 6.507159 12.296025 14.287846 7.503581 23.035727
#> [5656] 44.507807 21.540643 29.158486 8.421196 14.577786
#> [5661] 16.518687 6.551813 17.652274 40.726932 21.477399
#> [5666] 27.832757 5.174307 17.541639 15.466425 6.045972
#> [5671] 23.942745 42.047886 20.552341 21.297594 11.327878
#> [5676] 14.112659 13.465211 7.561116 20.891421 39.545105
#> [5681] 19.466234 25.902088 6.970867 10.721802 14.237479
#> [5686] 7.934469 18.993176 43.471168 18.829538 28.251999
#> [5691] 6.619835 14.278923 11.615486 10.125237 25.820873
#> [5696] 37.290990 22.039825 45.526493 8.860670 13.170684
#> [5701] 11.965083 5.701149 22.970239 47.536575 17.859171
#> [5706] 27.403849 11.756828 13.365287 11.563088 6.603249
#> [5711] 19.394457 38.509610 19.594411 27.302006 7.978791
#> [5716] 12.335761 13.157835 5.887464 20.281194 50.624110
#> [5721] 18.197701 23.702696 12.613101 13.236488 15.888507
#> [5726] 6.469216 20.713158 33.497166 21.683398 20.855007
#> [5731] 6.527923 14.409809 12.152113 5.829081 21.829375
#> [5736] 42.967839 18.284162 25.953627 7.302648 15.312105
#> [5741] 11.163051 7.059352 22.451720 55.118277 14.706257
#> [5746] 24.231435 6.239305 11.933300 11.482309 13.922378
#> [5751] 23.312185 35.999993 18.468304 22.806715 6.053295
#> [5756] 13.681849 10.199348 6.919740 21.173732 38.787369
#> [5761] 18.394192 24.134528 5.934901 11.611764 11.985345
#> [5766] 6.137566 25.370443 36.355738 30.076121 24.597549
#> [5771] 8.476769 11.829287 12.237094 7.833082 17.024092
#> [5776] 31.942338 17.740252 26.062882 7.070393 13.139167
#> [5781] 10.057769 5.095920 20.020777 40.341561 15.354534
#> [5786] 29.237354 8.457130 10.475425 10.036429 10.087661
#> [5791] 21.093659 47.750642 17.410353 29.669635 6.080444
#> [5796] 12.988895 10.065906 7.317907 19.790424 30.071568
#> [5801] 26.804742 22.730861 6.864117 10.676785 11.237936
#> [5806] 5.643706 21.637751 43.567709 27.582795 24.688186
#> [5811] 6.651049 12.489531 12.305251 5.027223 21.187252
#> [5816] 39.480930 17.954805 21.496859 7.088664 10.972968
#> [5821] 10.433393 7.064342 17.486392 41.079621 12.462918
#> [5826] 22.563665 4.645000 10.210050 9.841871 4.545358
#> [5831] 18.956592 37.958999 17.593745 23.096646 5.675881
#> [5836] 11.308275 11.962768 6.282951 20.170424 47.077768
#> [5841] 15.466341 23.863601 7.573546 11.824666 10.677047
#> [5846] 4.094874 19.369373 48.449219 16.441603 28.571513
#> [5851] 5.143756 13.317933 10.858247 6.331893 21.963200
#> [5856] 36.229585 15.474986 20.541464 7.247845 12.542505
#> [5861] 11.423010 6.824097 15.968173 39.267411 14.246214
#> [5866] 23.859755 4.786252 10.830720 9.510862 4.685188
#> [5871] 17.486187 32.752970 14.303565 21.148966 7.471369
#> [5876] 11.557634 9.989837 5.051698 19.569410 42.029570
#> [5881] 15.710827 20.697971 6.101037 10.979036 12.173694
#> [5886] 5.356524 18.635946 34.636425 16.274177 22.425319
#> [5891] 7.265970 11.199063 11.075436 5.460401 16.274875
#> [5896] 43.487680 16.894735 22.490648 5.658307 11.431265
#> [5901] 12.308903 3.716979 14.761261 30.137723 26.085530
#> [5906] 23.743736 5.511207 9.487293 11.727344 3.946854
#> [5911] 18.358340 30.042680 17.640732 19.396595 5.540704
#> [5916] 12.829256 9.375596 4.478811 20.907189 31.625138
#> [5921] 14.311768 21.888127 6.201748 10.436704 11.062678
#> [5926] 4.800994 22.197087 37.329104 16.905886 21.342389
#> [5931] 4.184414 9.794905 12.300346 6.322615 19.054385
#> [5936] 25.624018 15.559821 21.976108 5.925293 10.548200
#> [5941] 11.061320 3.928094 21.721847 31.082930 15.730390
#> [5946] 22.552142 4.821667 11.816226 11.516344 5.600983
#> [5951] 19.042766 27.369907 13.867009 19.324885 4.316336
#> [5956] 11.744548 10.201385 6.862863 14.868542 25.671994
#> [5961] 21.497115 19.950111 5.227230 12.352900 11.793891
#> [5966] 7.922085 16.467514 29.233923 19.342515 24.204033
#> [5971] 6.817222 11.201137 13.313019 4.572647 24.195375
#> [5976] 31.381669 17.205520 22.203010 11.207973 10.988817
#> [5981] 8.733691 4.249936 22.672829 27.506185 18.057019
#> [5986] 22.534012 5.698990 10.988898 12.000976 5.925525
#> [5991] 17.584087 36.361166 17.059897 19.463759 5.744939
#> [5996] 14.029562 12.062251 6.391228 20.279620 20.916822
#> [6001] 15.465160 21.549043 6.722870 12.028100 9.949838
#> [6006] 6.007326 24.382272 33.732603 18.271602 21.577365
#> [6011] 6.615802 10.172838 10.803664 3.854277 18.433202
#> [6016] 30.541047 18.613627 21.680726 7.294082 11.106864
#> [6021] 12.282757 5.239440 19.904770 28.836984 15.674173
#> [6026] 21.713432 13.651867 11.300693 10.528669 7.567143
#> [6031] 18.464297 29.124795 14.150920 25.964121 5.663315
#> [6036] 11.118359 11.280843 5.713714 17.904503 36.231586
#> [6041] 21.510322 18.696684 5.041147 9.124287 11.431144
#> [6046] 3.798698 18.065674 25.423382 20.677185 20.894602
#> [6051] 5.702843 10.777248 10.712305 5.173653 20.071802
#> [6056] 36.382167 17.871196 22.533954 6.061124 11.017222
#> [6061] 9.661962 5.581918 18.825902 30.713348 22.574746
#> [6066] 21.728186 6.972377 11.105360 13.780225 4.550884
#> [6071] 19.395026 26.880207 14.210696 22.155957 6.797550
#> [6076] 10.275557 12.222819 8.398578 24.344253 34.193368
#> [6081] 15.504388 20.300883 5.677231 10.249366 12.681867
#> [6086] 4.815232 26.414719 32.685804 19.477807 25.813704
#> [6091] 5.742608 10.393623 11.986271 6.906906 16.276390
#> [6096] 26.596281 13.400807 18.981896 4.858285 10.454178
#> [6101] 12.701189 7.966966 20.260167 30.027440 18.859080
#> [6106] 22.618743 5.682270 12.054238 11.638004 5.021385
#> [6111] 15.539340 34.765216 19.191591 15.998954 5.048621
#> [6116] 9.386482 13.078936 5.873381 15.623255 28.514973
#> [6121] 15.029830 19.101041 6.477837 11.175430 12.146461
#> [6126] 5.148064 18.652995 29.943941 18.033890 23.966590
#> [6131] 6.252118 9.911543 11.501488 3.347896 14.578482
#> [6136] 25.920330 16.644279 17.580480 6.416454 10.302214
#> [6141] 9.645012 6.561954 17.809095 26.193635 16.791367
#> [6146] 25.535732 4.844136 11.837402 8.871229 4.757383
#> [6151] 15.651360 26.396547 14.027239 19.791865 9.792290
#> [6156] 8.804900 10.444221 5.243068 19.937082 26.089301
#> [6161] 18.055032 22.551599 6.192553 9.915538 9.173633
#> [6166] 5.868727 15.541182 24.232333 14.435194 23.942167
#> [6171] 5.017874 10.179691 11.269468 4.221513 20.727558
#> [6176] 26.321338 19.164747 18.945027 7.669707 11.056863
#> [6181] 11.127577 12.666749 15.256717 25.997107 17.438546
#> [6186] 19.986527 6.671356 10.993758 9.369533 4.444676
#> [6191] 21.731148 28.489718 13.770881 18.928200 5.659063
#> [6196] 10.686092 9.026780 4.218146 15.360442 32.986778
#> [6201] 14.450117 23.222753 6.086246 10.253449 10.288777
#> [6206] 5.155154 16.632369 24.452634 15.953815 16.944394
#> [6211] 5.786364 10.837662 12.010598 4.611328 19.639332
#> [6216] 23.316960 16.959275 25.506402 4.360476 10.334268
#> [6221] 11.445835 8.239900 14.574909 25.082319 11.856776
#> [6226] 26.322803 4.254589 11.393935 9.486259 4.610584
#> [6231] 15.952926 27.274178 16.735782 21.962689 5.648849
#> [6236] 12.947390 11.642464 4.671631 15.635235 25.916409
#> [6241] 16.055308 18.728775 6.844440 9.780797 10.753023
#> [6246] 6.802180 21.511615 52.861304 13.085859 21.294226
#> [6251] 5.747175 8.597011 10.058490 8.907762 15.482151
#> [6256] 35.679026 16.521257 27.283316 4.951118 10.781809
#> [6261] 9.012623 6.569064 19.638396 29.239237 13.164390
#> [6266] 23.731186 5.510069 10.607306 13.007705 4.549610
#> [6271] 18.385448 33.727136 14.876893 28.768128 6.202144
#> [6276] 11.445276 11.072589 5.572842 20.476118 25.933766
#> [6281] 15.658919 20.068896 5.536041 10.364386 12.191351
#> [6286] 4.242294 18.140369 28.400607 17.945117 23.952858
#> [6291] 5.254488 9.975148 13.029144 4.996982 16.987497
#> [6296] 21.594442 19.360542 18.129262 6.440756 9.416642
#> [6301] 8.879312 3.974525 22.951163 29.104788 17.868570
#> [6306] 20.328365 11.548576 10.085428 10.498596 4.715607
#> [6311] 24.529314 26.396005 21.096141 19.854503 9.045296
#> [6316] 10.613922 12.959016 5.471734 21.122166 30.976210
#> [6321] 17.467314 30.765345 6.214583 10.425230 9.409511
#> [6326] 5.592456 16.731984 23.036097 17.275026 21.311622
#> [6331] 5.486886 10.735616 9.364954 3.541165 20.435235
#> [6336] 23.682017 12.751667 19.746048 7.011281 9.291310
#> [6341] 8.632754 4.888646 22.854153 42.580133 22.856214
#> [6346] 20.414992 6.439880 11.011290 12.585844 4.953826
#> [6351] 24.834518 30.640564 13.469658 30.635091 6.262156
#> [6356] 11.240475 11.193622 4.392331 19.049030 28.331619
#> [6361] 19.054333 33.136650 5.985691 10.087040 9.629414
#> [6366] 4.252129 17.535644 22.333125 15.536449 20.266349
#> [6371] 5.705797 11.686510 12.066039 6.810281 21.422717
#> [6376] 30.576053 15.644490 18.046062 6.146557 11.416335
#> [6381] 11.477552 5.818372 18.691383 21.365292 13.661480
#> [6386] 19.384176 5.654201 11.051018 10.236746 5.007565
#> [6391] 18.888312 25.496562 12.647561 24.443660 7.740616
#> [6396] 11.405505 11.184417 12.563951 20.612560 27.299467
#> [6401] 16.240234 21.575600 6.522759 10.468161 11.962806
#> [6406] 4.640039 12.203463 20.964559 15.498840 19.165081
#> [6411] 4.929392 10.422037 11.032308 7.748884 19.017662
#> [6416] 28.355905 13.492469 20.585249 6.358037 10.892174
#> [6421] 10.782553 6.424071 20.991071 26.787088 36.758570
#> [6426] 21.156857 5.091073 10.649386 10.809779 5.820364
#> [6431] 18.408146 35.748520 25.736241 19.774131 7.259056
#> [6436] 10.382411 10.373931 4.647639 14.995460 22.978675
#> [6441] 19.219628 22.706139 7.141451 11.857613 10.200689
#> [6446] 5.671289 20.476081 25.707751 17.518600 30.063380
#> [6451] 7.862410 11.423195 12.148871 6.945512 16.150527
#> [6456] 24.298117 14.559476 20.360127 4.588653 11.398603
#> [6461] 9.113533 4.713270 16.622362 31.611712 14.491402
#> [6466] 21.475547 6.315935 11.508246 11.642655 5.202209
#> [6471] 20.508794 30.428672 19.069939 21.935285 6.848964
#> [6476] 11.256043 12.959254 11.712290 17.863851 23.550501
#> [6481] 12.352790 26.415876 5.366100 11.636339 11.313623
#> [6486] 4.908458 18.968952 26.960806 18.474412 23.130283
#> [6491] 6.378123 11.770825 9.588354 9.072638 18.655192
#> [6496] 26.455795 21.032671 31.279063 7.377360 12.570899
#> [6501] 9.846947 4.500947 18.131702 24.298941 17.305334
#> [6506] 20.882561 6.153599 9.618351 10.714424 11.896306
#> [6511] 20.155444 29.094227 15.572138 28.487446 10.424288
#> [6516] 11.914281 12.415575 5.900483 19.919316 22.812075
#> [6521] 15.593424 26.030601 7.840797 11.509692 12.017372
#> [6526] 8.057531 22.592977 22.892289 14.215761 20.443693
#> [6531] 5.393908 11.870577 11.834267 6.053441 15.883934
#> [6536] 23.083281 16.238323 22.422582 6.289417 10.830067
#> [6541] 10.223057 7.172563 17.810912 24.632146 13.679702
#> [6546] 20.500806 6.857304 10.062145 9.713911 6.287977
#> [6551] 14.404737 27.440468 17.232877 26.050061 16.110817
#> [6556] 9.997268 11.447893 4.527345 17.506791 27.246427
#> [6561] 15.694653 20.233884 6.666143 8.928364 10.965052
#> [6566] 3.648313 23.213616 33.352166 15.071790 36.678364
#> [6571] 7.022363 11.270742 8.838623 13.090240 19.128171
#> [6576] 35.436642 19.507580 18.148779 4.615980 9.578203
#> [6581] 10.253686 4.703976 20.169298 22.556414 16.640852
#> [6586] 24.354504 5.193703 12.812498 10.455099 4.696572
#> [6591] 22.974636 27.090029 19.984680 25.796121 7.334822
#> [6596] 11.942260 10.560468 5.169543 20.273639 26.778747
#> [6601] 17.712928 47.051205 5.623750 10.359454 10.664079
#> [6606] 4.895109 19.253780 27.860563 17.694158 28.949455
#> [6611] 8.289831 11.102530 11.607740 3.466563 14.714239
#> [6616] 29.265666 14.398066 22.644254 5.936996 10.778961
#> [6621] 11.092141 4.643042 21.476321 27.218363 17.894117
#> [6626] 23.355493 5.036956 9.768475 10.394807 4.895581
#> [6631] 20.026397 29.728378 14.898927 20.384213 5.368960
#> [6636] 10.511783 10.439251 6.390248 22.113762 28.523886
#> [6641] 13.802355 21.631001 9.026024 11.403928 9.918543
#> [6646] 4.623812 15.742373 28.029476 18.554355 22.773872
#> [6651] 5.746443 11.767112 10.083992 4.374796 17.626457
#> [6656] 27.991132 15.331903 23.400512 6.216858 12.313058
#> [6661] 10.803967 5.525187 15.587872 23.094022 15.856851
#> [6666] 18.961042 6.198267 11.592470 10.174680 4.796777
#> [6671] 19.315707 27.416641 15.184901 24.891714 6.427024
#> [6676] 10.255672 11.831430 5.093051 20.173487 27.496317
#> [6681] 20.103094 38.396415 6.865835 11.102031 11.310023
#> [6686] 5.478815 18.663656 24.478150 16.901445 21.618265
#> [6691] 7.999599 10.073415 7.744202 8.749388 19.594045
#> [6696] 26.903182 14.525386 22.084893 5.142531 11.084978
#> [6701] 10.620466 7.738099 18.321208 29.081020 13.301647
#> [6706] 19.992725 5.895386 10.644138 11.859441 7.282677
#> [6711] 18.018021 28.188249 24.760736 22.038372 4.366667
#> [6716] 11.001688 12.499550 5.123093 15.262302 23.756095
#> [6721] 20.720150 30.228790 4.394520 10.028479 9.707609
#> [6726] 4.350069 18.140195 20.009206 17.993648 19.002447
#> [6731] 6.299981 11.473525 9.543563 4.155215 15.328477
#> [6736] 28.991882 17.768750 22.615224 6.139764 11.501982
#> [6741] 13.734892 5.499403 16.914373 28.790825 16.872875
#> [6746] 18.492110 5.583537 8.935665 11.769684 5.286785
#> [6751] 18.034496 32.084175 15.515344 19.329474 8.072934
#> [6756] 11.121037 9.593857 5.412797 19.771129 23.106614
#> [6761] 17.579322 20.187170 5.363943 11.451182 11.587418
#> [6766] 5.364188 16.048256 25.325948 15.721866 18.614385
#> [6771] 6.805010 9.811136 11.751012 3.874564 23.567623
#> [6776] 23.543039 13.171079 27.936709 8.197528 11.255197
#> [6781] 8.107489 7.452791 21.564299 27.342121 12.945750
#> [6786] 25.453192 6.986556 9.905569 9.395154 4.332932
#> [6791] 16.783253 22.894339 24.797071 22.335369 6.891753
#> [6796] 11.678509 10.689191 4.912379 21.466754 23.509351
#> [6801] 13.218125 17.953736 8.571553 9.593539 10.262257
#> [6806] 6.482088 16.753287 34.789242 15.390476 22.116460
#> [6811] 6.477632 10.788972 13.416375 5.291164 21.663222
#> [6816] 32.065756 14.023479 30.832228 5.314142 9.250391
#> [6821] 9.214860 6.348743 18.339258 24.655968 13.249055
#> [6826] 55.532747 6.780604 11.377478 13.323924 6.216104
#> [6831] 14.935224 25.768693 12.909203 22.671930 4.488649
#> [6836] 10.799970 10.937674 5.255813 19.616507 23.172698
#> [6841] 15.823211 21.245194 7.096178 9.550661 11.152417
#> [6846] 8.152236 21.084419 26.770305 14.429645 21.271298
#> [6851] 7.832856 12.056087 11.779300 4.475797 18.643286
#> [6856] 26.387191 14.730316 36.594233 8.089238 11.029222
#> [6861] 10.422804 7.267043 15.095851 26.593623 21.754364
#> [6866] 23.551933 5.089353 10.360865 11.938029 5.753962
#> [6871] 20.659532 24.610320 21.376482 20.406358 5.275592
#> [6876] 9.250746 10.613810 3.960601 17.330385 30.206389
#> [6881] 15.360810 29.016053 5.530230 11.171647 9.412464
#> [6886] 5.113704 21.928675 38.104277 11.901806 21.775210
#> [6891] 5.200633 10.539019 10.844053 6.163663 16.175943
#> [6896] 36.678962 16.282467 38.056857 5.362595 11.323132
#> [6901] 10.703461 5.397676 15.702316 24.359320 15.139917
#> [6906] 18.602421 4.762504 10.856186 14.184799 8.808151
#> [6911] 14.704503 31.305581 11.804618 26.150565 6.763011
#> [6916] 9.497258 11.582165 5.394998 23.759381 20.320550
#> [6921] 15.241552 16.246039 4.684850 10.979298 10.456820
#> [6926] 8.905588 14.781592 27.012876 14.292981 33.032637
#> [6931] 5.100140 10.400169 13.934594 4.795497 17.293917
#> [6936] 20.621456 13.904029 17.864621 5.156797 10.480541
#> [6941] 10.470159 4.898430 14.563778 25.385020 21.642064
#> [6946] 18.811826 4.960947 11.097639 13.182356 5.667703
#> [6951] 18.891917 21.366534 12.954209 22.733060 7.033172
#> [6956] 11.417416 9.605428 5.253914 19.270777 25.843744
#> [6961] 14.485716 25.597451 5.316602 10.640504 9.586580
#> [6966] 5.366495 13.176101 30.195574 18.606871 17.343704
#> [6971] 7.126794 9.636645 12.942500 4.748871 18.303082
#> [6976] 23.545868 16.706177 25.737615 6.227998 11.098120
#> [6981] 10.200699 6.050280 18.225609 27.069885 14.260101
#> [6986] 19.685704 5.140146 12.979811 9.685239 3.892039
#> [6991] 19.996609 28.714788 12.774022 20.914691 5.563148
#> [6996] 11.257266 7.818056 8.191035 19.670319 24.455115
#> [7001] 13.901029 20.666881 5.740455 10.390315 10.540737
#> [7006] 5.239556 18.002698 30.751921 14.245768 22.747782
#> [7011] 6.905547 11.496915 12.349256 6.612709 18.463281
#> [7016] 28.900459 15.671822 19.157494 7.323224 10.722297
#> [7021] 11.034751 10.277040 21.406327 29.377878 14.209451
#> [7026] 20.876081 5.839155 11.080671 10.919747 5.575396
#> [7031] 15.760910 26.419115 13.416710 16.347512 5.374161
#> [7036] 9.481983 12.547474 4.060674 17.018011 30.737735
#> [7041] 13.810171 17.649129 5.622687 10.487182 10.083001
#> [7046] 6.024245 15.634883 30.548391 25.156382 29.441643
#> [7051] 8.865323 12.989413 12.099445 4.575110 15.880972
#> [7056] 28.032077 15.624855 26.689582 7.208590 9.815443
#> [7061] 10.065303 3.596404 18.388266 28.468746 15.937781
#> [7066] 18.162128 6.642714 10.818792 13.257751 3.985955
#> [7071] 15.419146 26.977868 13.988382 17.933015 8.956976
#> [7076] 10.258652 12.037752 3.863613 18.960030 32.534989
#> [7081] 14.185539 16.000703 6.372031 11.121082 12.152146
#> [7086] 5.688608 17.753208 30.389682 15.119966 19.452684
#> [7091] 8.024150 10.194372 10.160811 7.108232 17.640853
#> [7096] 25.193212 13.075439 16.072879 6.290957 9.592162
#> [7101] 12.689229 4.033864 20.406621 23.419240 18.145626
#> [7106] 21.302784 5.481212 11.696416 11.580833 4.831010
#> [7111] 22.675253 32.060353 16.695246 25.414814 6.109536
#> [7116] 11.846644 10.857186 5.205461 16.780919 25.427510
#> [7121] 16.107229 17.122453 5.059080 10.556342 11.822079
#> [7126] 4.515281 14.988842 30.244731 12.690808 19.665574
#> [7131] 6.713899 9.560162 9.421713 5.681562 16.055999
#> [7136] 28.171817 20.712684 20.664815 6.748661 12.571693
#> [7141] 8.823148 5.822341 20.121793 20.979256 18.631864
#> [7146] 21.673262 7.629184 13.502819 9.073333 8.684805
#> [7151] 16.186168 21.464304 15.091964 19.862444 4.927465
#> [7156] 12.930948 14.048988 7.158971 17.242263 23.563460
#> [7161] 15.466045 19.277646 7.587924 10.872017 9.533815
#> [7166] 4.970147 16.913882 24.449818 43.758709 25.863644
#> [7171] 5.232467 9.738889 11.804419 8.489460 16.669560
#> [7176] 26.688257 17.219719 27.996507 5.178023 11.167806
#> [7181] 11.004149 4.800555 19.655563 43.036808 15.986097
#> [7186] 18.054808 5.628900 11.407267 10.183817 4.532392
#> [7191] 20.219760 27.134075 13.048674 21.588818 5.518193
#> [7196] 12.782701 9.393076 5.146381 18.904115 22.286011
#> [7201] 18.671808 24.223806 8.796984 11.846353 13.320911
#> [7206] 6.489286 19.517095 29.724709 10.712072 17.545316
#> [7211] 4.368678 8.627120 10.559412 5.426895 17.799580
#> [7216] 25.874331 17.997040 18.517430 5.618141 11.743682
#> [7221] 11.158027 3.575223 20.657490 32.562441 19.037497
#> [7226] 26.273791 6.283998 12.640235 11.624089 4.913197
#> [7231] 20.571886 30.399636 15.870522 24.490590 6.410994
#> [7236] 13.209832 11.364761 5.087315 16.052558 22.931022
#> [7241] 16.044677 31.339568 6.483887 11.570144 10.642505
#> [7246] 11.810883 20.532468 24.897924 14.516260 19.030195
#> [7251] 5.798249 10.029972 9.873241 3.567322 15.658822
#> [7256] 23.023953 13.123244 23.926514 4.117592 10.599039
#> [7261] 10.300942 3.321613 20.175308 23.785457 14.509117
#> [7266] 19.682312 4.608440 10.591288 9.225198 8.423284
#> [7271] 22.571393 22.472397 14.747287 19.000214 5.375725
#> [7276] 11.626566 13.137880 4.952970 19.718954 26.383357
#> [7281] 15.215226 28.117391 5.542641 12.041772 11.480610
#> [7286] 5.272277 20.217408 34.523173 15.138853 27.339873
#> [7291] 4.612850 11.663930 10.295282 5.648234 16.710742
#> [7296] 26.140353 16.727275 22.147068 5.179146 10.149677
#> [7301] 10.860208 4.961367 18.097953 27.945734 18.120679
#> [7306] 21.437620 6.141739 10.819232 9.853445 5.869381
#> [7311] 15.428073 27.692158 19.264237 27.566039 6.393489
#> [7316] 10.265059 10.295941 3.146296 14.911231 27.209805
#> [7321] 17.108452 22.307817 5.526573 11.341389 10.502163
#> [7326] 5.046900 23.287188 26.458952 18.182431 15.872881
#> [7331] 4.926028 10.896387 9.224376 6.180670 17.732998
#> [7336] 21.434360 18.239937 20.242460 6.465121 12.215855
#> [7341] 10.073714 7.710437 19.436794 34.905662 18.347346
#> [7346] 24.781970 4.619737 10.903837 9.441030 2.893308
#> [7351] 16.659490 39.575581 14.102785 22.814500 5.316469
#> [7356] 13.326233 10.705395 5.415616 20.063398 23.376480
#> [7361] 17.464293 21.574140 4.836159 11.063343 9.073998
#> [7366] 5.139404 20.440055 32.329492 18.073725 24.193438
#> [7371] 5.621191 11.747614 10.044469 4.964368 21.480612
#> [7376] 24.070078 18.499838 25.460212 5.770030 11.065400
#> [7381] 11.097140 5.494908 20.947693 20.181266 14.691009
#> [7386] 18.358518 12.839608 10.779411 11.778241 4.042762
#> [7391] 14.593209 24.424841 18.810647 30.670111 5.781123
#> [7396] 12.204886 12.370435 7.702345 19.604917 27.572259
#> [7401] 14.847023 22.105461 5.941954 11.608932 11.650530
#> [7406] 4.810545 15.205574 27.321689 15.771732 29.597042
#> [7411] 5.330957 10.728306 11.132029 11.893980 19.501164
#> [7416] 25.440793 11.841345 18.703986 5.092588 13.080661
#> [7421] 11.049942 13.596787 16.725051 28.551876 18.751091
#> [7426] 20.968913 5.036960 10.868268 11.691249 5.074553
#> [7431] 19.074393 20.464564 13.393238 22.127389 5.214133
#> [7436] 11.972148 10.633052 5.715829 17.524953 30.918745
#> [7441] 17.044198 20.282594 5.437108 12.423008 11.990285
#> [7446] 4.790931 19.447584 29.974709 19.251738 17.455773
#> [7451] 6.751485 11.323130 10.531420 7.612166 21.257715
#> [7456] 20.800256 13.720112 25.410774 4.919325 10.800045
#> [7461] 9.372462 5.072446 20.185149 38.150772 19.376895
#> [7466] 20.854240 5.337673 11.381572 12.616399 3.990814
#> [7471] 18.291332 25.068256 12.631867 23.330574 6.869057
#> [7476] 12.553170 11.815984 5.021365 20.802859 28.155576
#> [7481] 15.764191 26.821764 6.360442 11.766035 10.782271
#> [7486] 3.896033 17.382828 27.710311 18.100761 17.079317
#> [7491] 7.684174 11.574225 10.768669 8.289828 18.917753
#> [7496] 26.804411 13.688045 21.537911 5.078491 12.180277
#> [7501] 10.125505 5.288098 22.262421 24.846690 21.931702
#> [7506] 23.686373 6.074599 11.436642 10.600646 4.518934
#> [7511] 21.997900 21.524193 23.527411 26.759419 7.094081
#> [7516] 12.090066 10.643323 4.132045 18.537347 19.476560
#> [7521] 25.686040 21.352789 6.128181 12.199460 13.321196
#> [7526] 2.734434 21.206933 23.053278 17.372551 25.798210
#> [7531] 9.698968 14.378929 12.645599 7.810056 20.034915
#> [7536] 32.555793 16.791938 25.004879 5.251820 13.702863
#> [7541] 10.538479 5.009352 22.555193 19.712689 26.936604
#> [7546] 17.422569 6.176965 14.143801 12.003666 3.828041
#> [7551] 17.644515 25.042951 16.887221 30.769232 9.324177
#> [7556] 12.406835 12.606379 5.283516 18.386701 29.474921
#> [7561] 15.251721 19.601222 10.085241 9.814753 10.810465
#> [7566] 4.140590 19.163609 20.468277 14.263511 22.320248
#> [7571] 5.150916 10.569625 11.878047 6.988212 17.878611
#> [7576] 26.540782 15.207089 20.221743 14.457465 11.367417
#> [7581] 11.208363 4.907654 21.556411 30.568173 14.046807
#> [7586] 19.669251 6.402272 13.619962 10.007125 5.780530
#> [7591] 23.186570 27.099030 15.929814 31.258148 7.509852
#> [7596] 9.551942 13.551723 5.922459 16.579424 20.662296
#> [7601] 19.112092 25.091589 4.978148 11.568894 12.625133
#> [7606] 4.354787 23.936968 26.683195 14.458707 21.009509
#> [7611] 12.612336 9.856103 11.225819 8.167260 16.837622
#> [7616] 20.325522 16.058894 20.026479 5.190318 12.508379
#> [7621] 11.215948 4.217643 19.692705 29.805327 15.671152
#> [7626] 18.085552 5.073741 12.106583 11.490157 5.347962
#> [7631] 18.824891 49.726941 14.281069 20.817688 8.415164
#> [7636] 9.796088 10.595431 5.451450 16.636279 31.143978
#> [7641] 19.292997 16.612393 4.891810 11.551406 12.025671
#> [7646] 3.924839 19.266377 22.736391 16.075325 30.839272
#> [7651] 5.590718 13.171760 11.567755 6.094387 16.769438
#> [7656] 28.060955 12.635570 18.827009 4.027460 12.082643
#> [7661] 9.984518 4.672091 24.819565 30.471918 16.111701
#> [7666] 22.382919 4.645265 12.025229 13.000555 5.091949
#> [7671] 26.920995 26.348043 19.936282 24.945016 6.827643
#> [7676] 11.023471 10.900113 8.315678 21.473304 27.530558
#> [7681] 12.454847 21.831697 5.573722 10.601059 11.925730
#> [7686] 5.746961 20.713728 25.798588 16.012434 21.796169
#> [7691] 6.417882 10.700514 10.903377 4.480478 17.149674
#> [7696] 23.878620 18.499575 25.481645 6.656719 13.782775
#> [7701] 12.623189 5.521048 25.624412 31.446268 16.421929
#> [7706] 20.429872 4.733633 10.520906 11.185344 4.420251
#> [7711] 22.497928 36.608600 15.347308 19.480339 7.083301
#> [7716] 10.731928 14.869575 5.547368 18.916802 25.658793
#> [7721] 15.953297 25.185723 7.185226 12.205838 11.457208
#> [7726] 8.220383 17.299302 23.415723 14.061044 20.435312
#> [7731] 7.046283 12.713612 9.526309 7.821084 19.330632
#> [7736] 30.619985 12.920738 25.131478 6.298253 11.351445
#> [7741] 11.885363 4.346974 20.596821 34.553287 15.620047
#> [7746] 18.851762 3.972778 13.309963 7.983911 4.375903
#> [7751] 17.639627 30.102932 111.255685 107.742982 39.323458
#> [7756] 90.245908 41.333908 34.393383 60.231789 1347.062580
#> [7761] 258.993888 149.097302 80.886770 183.057364 380.588923
#> [7766] 221.053078 231.076300 2751.860237 427.376555 220.757567
#> [7771] 181.393390 275.608580 891.967851 372.613964 324.520833
#> [7776] 2654.088363 605.403913 261.982388 205.642529 331.290232
#> [7781] 737.029419 389.073368 315.570563 2277.503047 615.309312
#> [7786] 300.432175 252.564125 404.578241 908.647718 649.023116
#> [7791] 423.792728 1707.197354 827.826970 349.325771 352.824885
#> [7796] 457.667557 702.284586 429.008773 344.530821 2202.876764
#> [7801] 768.317060 465.955844 290.023176 385.290865 623.483698
#> [7806] 402.191514 305.123658 1714.732878 588.782495 461.701825
#> [7811] 247.541897 320.326130 489.561579 343.556420 303.208162
#> [7816] 1539.537585 651.452719 378.687139 248.363507 302.456540
#> [7821] 429.367335 317.360105 250.107659 1538.783435 509.186961
#> [7826] 391.489768 233.971815 260.377352 347.627022 264.328962
#> [7831] 207.426240 1365.394803 523.692695 327.306901 192.844284
#> [7836] 256.843326 362.948637 242.274251 193.427337 1348.491000
#> [7841] 479.303437 285.394727 165.414905 245.095873 336.400719
#> [7846] 217.795558 192.110785 1252.299473 468.573697 439.146607
#> [7851] 186.816167 218.680490 299.378622 222.052751 172.299356
#> [7856] 1210.765626 476.897979 355.980335 183.315618 236.212957
#> [7861] 271.984407 213.512437 180.891237 1179.405691 434.962204
#> [7866] 293.513382 183.294823 251.041310 263.574969 198.637262
#> [7871] 170.918953 1072.756967 440.406159 270.230960 185.138421
#> [7876] 219.393022 234.354640 167.557228 145.656116 1003.720204
#> [7881] 420.371526 280.616540 167.380158 204.790994 212.103685
#> [7886] 159.726511 136.725763 924.848811 454.933102 316.368792
#> [7891] 160.482277 185.209258 230.637496 156.850222 136.952103
#> [7896] 978.977708 408.307152 260.772104 162.477973 180.792609
#> [7901] 185.818480 126.458559 124.354105 924.736240 417.284080
#> [7906] 257.575590 154.447681 168.303761 162.235745 132.724399
#> [7911] 122.158344 858.140658 378.140355 305.451603 135.094730
#> [7916] 161.287850 158.749598 119.737923 117.369899 802.067620
#> [7921] 403.725657 232.105776 126.496602 159.704853 146.799901
#> [7926] 115.072374 103.631570 706.194285 334.775766 261.211185
#> [7931] 128.103187 149.964394 152.966866 117.347215 104.952157
#> [7936] 706.182840 344.046077 269.141127 123.395546 124.171349
#> [7941] 138.099311 82.074642 75.826916 589.043040 223.601279
#> [7946] 175.886064 142.444739 142.247683 111.930238 91.713923
#> [7951] 86.087973 595.800695 369.323350 212.722680 110.991849
#> [7956] 116.459330 129.801881 89.286497 82.584815 543.322802
#> [7961] 315.849776 218.248313 113.947610 121.421001 123.501765
#> [7966] 85.558560 82.558582 641.804290 345.811973 162.071403
#> [7971] 66.968154 141.820448 90.956242 80.273137 68.719788
#> [7976] 414.186888 184.411137 252.840866 110.139158 137.705735
#> [7981] 100.290282 50.685694 89.180058 497.646742 134.601899
#> [7986] 297.016990 85.664223 89.164163 61.245065 95.795605
#> [7991] 82.820765 534.519704 344.328371 196.745896 104.924516
#> [7996] 62.622743 113.532331 85.375663 82.439499 342.765803
## First we list available isolation windows
table(isolationWindowTargetMz(spectra(mse_dia)))
#>
#> 163.75 208.95 244.05 270.85 299.1 329.8 367.35 601.85
#> 1000 1000 1000 1000 1000 1000 1000 1000
## We can then extract the TIC of MS2 data for a specific isolation window
chr_ms2 <- chromatogram(mse_dia, msLevel = 2L,
isolationWindowTargetMz = 244.05)
plot(chr_ms2)
####
## Chromatographic peak detection
## Perform peak detection on the data using the centWave algorith. Note
## that the parameters are chosen to reduce the run time of the example.
p <- CentWaveParam(noise = 10000, snthresh = 40, prefilter = c(3, 10000))
xmse <- findChromPeaks(mse, param = p)
xmse
#> Object of class XcmsExperiment
#> Spectra: MS1 (3834)
#> Experiment data: 3 sample(s)
#> Sample data links:
#> - spectra: 3 sample(s) to 3834 element(s).
#> xcms results:
#> - chromatographic peaks: 248 in MS level(s): 1
## Have a quick look at the identified chromatographic peaks
head(chromPeaks(xmse))
#> mz mzmin mzmax rt rtmin rtmax into intb maxo
#> CP001 453.2 453.2 453.2 2506.073 2501.378 2527.982 1007409.0 1007380.8 38152
#> CP002 302.0 302.0 302.0 2617.185 2595.275 2640.659 687146.6 671297.8 30552
#> CP003 344.0 344.0 344.0 2679.783 2646.919 2709.517 5210015.9 5135916.9 152320
#> CP004 430.1 430.1 430.1 2681.348 2639.094 2712.647 2395840.3 2299899.6 65752
#> CP005 366.0 366.0 366.0 2679.783 2642.224 2718.907 3365174.0 3279468.3 79928
#> CP006 343.0 343.0 343.0 2678.218 2637.529 2712.647 24147443.2 23703761.7 672064
#> sn sample
#> CP001 38151 1
#> CP002 46 1
#> CP003 68 1
#> CP004 42 1
#> CP005 49 1
#> CP006 87 1
## Extract chromatographic peaks identified between 3000 and 3300 seconds
chromPeaks(xmse, rt = c(3000, 3300), type = "within")
#> mz mzmin mzmax rt rtmin rtmax into intb
#> CP021 453.2 453.2 453.2 3063.196 3035.027 3114.840 3001594.78 3001514.97
#> CP030 361.1 361.1 361.1 3147.704 3141.444 3153.964 367361.84 362278.28
#> CP031 340.3 340.3 340.3 3230.646 3225.952 3233.776 68671.25 68664.99
#> CP032 526.2 526.2 526.2 3243.166 3238.471 3246.296 374698.56 374692.30
#> CP033 313.1 313.1 313.1 3276.030 3254.121 3290.115 1199152.63 1162839.05
#> CP034 454.1 454.1 454.1 3276.030 3261.945 3294.809 12283448.95 12024951.77
#> CP108 307.1 307.1 307.1 3143.009 3121.099 3164.918 2191519.65 2063202.03
#> CP109 278.1 278.1 278.1 3196.217 3180.568 3213.432 657840.25 651048.36
#> CP110 526.1 526.1 526.1 3179.003 3150.833 3210.302 21334966.98 21147392.01
#> CP111 380.1 380.1 380.1 3152.398 3132.054 3174.308 2201517.91 2201474.09
#> CP112 380.1 380.1 380.1 3210.302 3210.302 3216.561 187031.44 187023.61
#> CP113 380.1 380.1 380.1 3202.477 3174.308 3216.561 1156780.73 1156736.91
#> CP114 380.1 380.1 380.1 3222.821 3216.561 3240.036 485818.25 485793.21
#> CP115 286.2 286.2 286.2 3258.815 3246.296 3280.725 1264118.64 1247233.70
#> CP116 308.1 308.1 308.1 3261.945 3241.601 3285.419 2066854.37 2025917.82
#> CP204 380.1 380.1 380.1 3150.835 3128.925 3171.179 2036803.99 1937655.24
#> CP205 286.2 286.2 286.2 3250.992 3233.777 3257.252 732016.23 722185.86
#> CP206 568.2 568.2 568.2 3207.173 3185.264 3232.212 3951832.25 3866503.74
#> maxo sn sample
#> CP021 53096 53095 1
#> CP030 49240 57 1
#> CP031 11391 11390 1
#> CP032 60800 60799 1
#> CP033 55392 50 1
#> CP034 554112 58 1
#> CP108 101400 43 2
#> CP109 30208 54 2
#> CP110 622144 251 2
#> CP111 95176 95175 2
#> CP112 25144 28791 2
#> CP113 28920 28919 2
#> CP114 23176 23535 2
#> CP115 67600 41 2
#> CP116 96272 78 2
#> CP204 82624 49 3
#> CP205 74824 68 3
#> CP206 164352 113 3
## Extract ion chromatograms (EIC) for the first two chromatographic
## peaks.
chrs <- chromatogram(xmse,
mz = chromPeaks(xmse)[1:2, c("mzmin", "mzmax")],
rt = chromPeaks(xmse)[1:2, c("rtmin", "rtmax")])
#> Extracting chromatographic data
#> Processing chromatographic peaks
## An EIC for each sample and each of the two regions was extracted.
## Identified chromatographic peaks in the defined regions are extracted
## as well.
chrs
#> XChromatograms with 2 rows and 3 columns
#> ko15.CDF ko16.CDF ko18.CDF
#> <XChromatogram> <XChromatogram> <XChromatogram>
#> [1,] peaks: 1 peaks: 0 peaks: 0
#> [2,] peaks: 1 peaks: 0 peaks: 0
#> phenoData with 3 variables
#> featureData with 4 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
## Plot the EICs for the second defined region
plot(chrs[2, ])
## Extracting MS2 chromatographic data
##
## To show how MS2 chromatograms can be extracted we first load a DIA
## (SWATH) data set.
library(MsDataHub)
mse_dia <- readMsExperiment(PestMix1_SWATH.mzML())
#> see ?MsDataHub and browseVignettes('MsDataHub') for documentation
#> loading from cache
## Extracting MS2 chromatogram requires also to specify the isolation
## window from which to extract the data. Without that chromatograms
## will be empty:
chr_ms2 <- chromatogram(mse_dia, msLevel = 2L)
intensity(chr_ms2[[1L]])
#> [1] 167.451218 105.432610 48.448413 42.587666 51.141832
#> [6] 32.010572 39.833950 162.208892 146.962103 116.968997
#> [11] 47.464513 43.562779 47.522917 32.687743 48.205317
#> [16] 154.777987 151.650104 114.490964 51.955748 40.298667
#> [21] 42.889420 26.168091 41.573644 160.917377 155.598118
#> [26] 115.631891 56.064858 40.531724 51.336031 35.671689
#> [31] 47.007723 176.209493 150.000631 129.402529 52.351775
#> [36] 41.211416 45.764487 30.722598 42.549656 162.606472
#> [41] 159.749701 131.409168 57.591062 40.961098 47.379330
#> [46] 35.990142 56.194426 176.991264 158.162751 128.185092
#> [51] 55.115210 38.549212 52.187233 29.735383 42.294920
#> [56] 168.506534 148.606744 123.903268 50.443994 37.206104
#> [61] 49.667910 36.124219 45.099398 160.804559 144.212288
#> [66] 117.309985 57.318721 40.807128 46.997778 31.888547
#> [71] 44.914434 162.760728 141.112480 116.353180 61.330481
#> [76] 42.391655 46.165895 36.830656 41.048551 173.389121
#> [81] 150.354805 121.448249 58.297946 39.415169 48.137585
#> [86] 33.945039 42.597284 176.705435 152.759067 117.964302
#> [91] 57.009782 41.762080 49.519787 33.609967 45.789184
#> [96] 173.561615 131.868021 118.099061 55.553144 39.780311
#> [101] 46.840269 34.194347 41.879404 157.716579 140.589185
#> [106] 121.374668 51.687723 40.195854 47.690116 37.602680
#> [111] 43.965245 156.025829 157.369269 110.587061 52.018941
#> [116] 39.871676 47.531537 33.916085 39.490682 162.735606
#> [121] 151.371999 115.130247 52.206799 41.082679 48.981736
#> [126] 29.885040 50.262584 174.824692 137.808708 118.701492
#> [131] 52.714717 36.914359 44.841750 35.428872 42.102767
#> [136] 176.969381 143.918775 122.172542 46.627960 39.409697
#> [141] 45.892751 32.955673 45.916826 164.074529 110.974725
#> [146] 121.420453 57.000711 38.071610 43.426923 31.968076
#> [151] 43.205524 167.359458 136.644147 114.657853 53.570604
#> [156] 37.251428 48.855616 28.654548 46.821310 159.257533
#> [161] 136.078653 122.850526 51.119067 36.936050 47.987294
#> [166] 31.154454 44.033667 187.065329 138.560486 121.384740
#> [171] 49.859420 41.431116 52.683159 31.240847 41.930264
#> [176] 166.090714 137.037807 126.818365 55.785933 40.353991
#> [181] 47.617070 32.448793 46.101182 178.091599 126.628694
#> [186] 125.285128 53.154098 38.196966 45.725605 33.988735
#> [191] 47.783238 176.030519 138.156590 121.158414 51.131236
#> [196] 37.260215 46.927999 32.118103 50.824118 163.126561
#> [201] 143.490280 119.269498 50.446773 39.403250 45.444075
#> [206] 31.371791 46.041094 163.995802 98.558906 97.119329
#> [211] 43.657266 31.607119 40.415288 19.480071 41.681597
#> [216] 165.475850 101.389368 103.396216 42.729989 20.902430
#> [221] 48.426218 26.930463 53.396440 187.151969 58.083847
#> [226] 108.166753 51.910657 24.056782 43.929434 29.711441
#> [231] 46.996525 143.340123 111.724883 130.201873 46.946882
#> [236] 30.703287 45.463666 25.532886 53.455083 188.459844
#> [241] 121.595032 128.265759 48.481593 36.367438 39.432143
#> [246] 35.110147 45.317950 199.461130 123.822349 124.453397
#> [251] 51.373002 40.373744 44.575361 34.098693 44.992004
#> [256] 199.302711 115.622113 115.520087 52.588235 36.559259
#> [261] 42.863897 19.107814 42.420217 216.812980 145.216247
#> [266] 118.223439 61.237153 31.971820 45.693190 30.493408
#> [271] 43.479256 214.007843 137.507353 135.967656 60.011754
#> [276] 37.082835 39.620731 30.998244 42.027486 232.012743
#> [281] 118.387136 142.364895 51.672463 38.945555 41.669065
#> [286] 33.612390 41.247462 196.533657 115.020205 142.988161
#> [291] 46.530636 36.358999 43.610314 34.859240 41.568579
#> [296] 199.727458 118.619273 127.635654 47.808546 34.739862
#> [301] 45.326667 38.867514 43.806293 202.771954 126.833096
#> [306] 164.186773 52.443501 41.062466 45.812602 36.456879
#> [311] 41.972819 222.238004 129.047394 146.835109 53.216151
#> [316] 36.054789 41.644602 41.153462 44.151703 198.428224
#> [321] 133.110851 127.782179 50.909340 39.017341 43.719600
#> [326] 42.062764 44.099596 187.029552 123.340463 116.238022
#> [331] 46.525230 41.095261 49.200918 45.152890 47.486162
#> [336] 226.301902 134.037386 124.916782 58.381726 40.923225
#> [341] 49.689228 36.423063 44.652538 161.173165 126.906557
#> [346] 103.237367 55.496240 42.352044 45.500991 34.862405
#> [351] 41.926576 163.237384 117.727915 105.705640 49.150506
#> [356] 38.811569 48.849277 27.522183 44.785886 160.226922
#> [361] 113.355423 112.975909 50.373055 39.141868 47.454134
#> [366] 31.329878 46.244481 158.126581 121.931115 118.672109
#> [371] 56.822938 37.047153 44.821043 35.484114 38.755738
#> [376] 191.300899 130.326700 117.394953 53.562705 36.812964
#> [381] 45.737268 31.711265 43.831297 158.679602 128.274776
#> [386] 111.081271 48.823742 39.079050 45.954053 31.563248
#> [391] 38.653086 176.238025 128.692405 118.366005 42.698888
#> [396] 39.021745 46.105397 35.345455 40.248080 175.256934
#> [401] 139.035411 112.353086 50.371917 37.354196 48.254171
#> [406] 32.303338 42.679164 171.239180 104.472487 122.455740
#> [411] 48.074323 38.934960 46.869484 35.927003 45.715158
#> [416] 228.863116 130.873426 123.462988 56.414827 41.828009
#> [421] 52.009153 37.171067 46.885994 241.372911 123.043119
#> [426] 133.669185 61.397251 41.912858 51.326151 34.145175
#> [431] 43.159684 193.986114 112.600631 120.207558 41.959882
#> [436] 33.638388 47.956467 32.989986 38.815009 182.225208
#> [441] 110.543328 126.495590 45.878389 30.322753 40.737404
#> [446] 27.611870 45.191360 162.945866 108.900070 118.947459
#> [451] 45.752574 29.518900 43.446930 28.691413 40.876603
#> [456] 152.296499 97.336724 116.545222 50.060893 34.709605
#> [461] 41.048271 35.368648 39.523235 152.532517 97.537269
#> [466] 92.912022 45.620119 29.189715 36.658149 26.662089
#> [471] 38.752345 128.852280 100.981794 109.578946 48.040893
#> [476] 34.831760 38.101074 28.549529 41.152690 141.402519
#> [481] 101.452419 113.073400 50.223908 32.221343 41.203864
#> [486] 30.189605 41.476329 150.812408 119.987223 126.791050
#> [491] 56.253979 33.970643 41.440763 28.418729 36.884502
#> [496] 151.521866 127.808146 107.266956 50.975376 35.513003
#> [501] 42.469854 30.469824 36.575157 145.856516 112.498257
#> [506] 97.783282 52.686416 34.647375 40.969349 27.201655
#> [511] 39.861164 147.592851 125.196846 107.886744 51.600730
#> [516] 37.969832 37.686576 33.473837 40.492766 138.681531
#> [521] 115.332075 116.144761 47.054372 33.797785 46.507380
#> [526] 27.791961 42.171336 140.036967 121.225005 104.036724
#> [531] 47.233675 38.214997 41.466050 32.206779 42.691704
#> [536] 139.523492 129.458122 111.883703 45.873956 34.418329
#> [541] 39.452800 28.672196 44.544770 132.852484 105.496340
#> [546] 98.788578 49.043451 33.354703 37.570898 28.401155
#> [551] 46.793168 124.081183 134.779078 111.072208 48.193504
#> [556] 37.352554 38.492679 30.140132 34.180987 150.349048
#> [561] 127.751322 109.830612 48.353302 33.361524 38.676967
#> [566] 27.177009 38.237094 151.832900 119.423590 108.777089
#> [571] 59.370222 27.981177 41.502313 28.070891 33.805593
#> [576] 153.805769 66.500519 100.033210 38.243472 21.429552
#> [581] 35.849464 27.832320 42.276980 141.855202 108.842237
#> [586] 113.209048 40.932875 31.415275 26.385132 25.796249
#> [591] 44.596828 136.734716 39.113411 88.657524 43.650184
#> [596] 30.565965 44.374236 25.166760 39.799153 139.989898
#> [601] 112.345237 105.048307 47.806467 34.410027 33.377036
#> [606] 25.100477 33.934663 134.232939 109.667560 108.007680
#> [611] 46.322747 31.528887 36.890234 28.285215 32.815617
#> [616] 129.861855 103.835167 105.723162 46.497357 27.632753
#> [621] 39.557204 28.456075 37.007709 122.480471 91.297184
#> [626] 99.087357 41.633584 31.281372 38.874662 30.189158
#> [631] 38.613619 133.617479 102.807951 97.771981 48.130536
#> [636] 30.429434 35.423973 26.915430 40.223614 139.184034
#> [641] 106.234774 104.388371 48.370299 31.882032 34.893013
#> [646] 25.255039 36.265976 129.206981 101.493353 99.002509
#> [651] 44.346640 32.158756 39.429191 26.705643 37.622683
#> [656] 132.237249 109.599833 92.242120 46.847184 31.010174
#> [661] 37.535520 27.512162 41.985617 137.100186 105.086888
#> [666] 94.188571 43.261979 33.923542 40.955904 24.140754
#> [671] 49.249031 145.383441 108.475989 102.639441 44.052413
#> [676] 31.671950 38.316922 26.313719 36.032487 145.339185
#> [681] 109.010582 111.256913 44.568090 32.622182 43.235431
#> [686] 28.846874 41.603560 185.257926 90.491924 55.381179
#> [691] 52.559675 35.404760 39.212934 26.798098 37.332781
#> [696] 207.062895 113.098940 112.052358 46.283524 25.168956
#> [701] 36.233487 27.672381 44.556306 179.957265 110.232423
#> [706] 102.707046 48.953326 36.362766 35.710726 34.303696
#> [711] 39.764515 188.588142 113.188109 118.948407 48.587346
#> [716] 33.652624 40.238380 31.320930 29.659764 191.976195
#> [721] 104.352510 105.337010 47.377342 30.138890 35.353116
#> [726] 30.443518 38.085864 161.939634 99.286794 113.933793
#> [731] 47.386050 31.276776 35.586038 28.099018 38.582196
#> [736] 158.502928 108.189300 108.556956 45.916943 29.664124
#> [741] 35.072582 29.660793 30.060255 163.596847 104.458447
#> [746] 111.339333 42.839141 30.381518 32.467032 27.207105
#> [751] 31.829775 152.395823 92.090382 101.389516 38.136271
#> [756] 30.162735 34.126044 27.465397 32.316456 159.614114
#> [761] 97.103220 93.203515 44.641377 27.876265 36.166051
#> [766] 25.380971 33.702906 155.271182 102.884171 96.148820
#> [771] 44.848030 30.642579 37.470218 27.501099 30.589302
#> [776] 163.899956 101.194633 95.039853 45.704995 32.728440
#> [781] 33.720749 26.186973 36.896967 156.103823 113.336619
#> [786] 98.596145 48.631773 33.012071 38.892305 28.817390
#> [791] 39.441818 171.890864 112.334461 107.901088 46.708054
#> [796] 32.391567 29.360276 26.269499 32.755082 160.337011
#> [801] 113.112331 96.390912 47.874924 32.144085 34.075555
#> [806] 32.297730 35.310021 149.723302 104.396845 99.148335
#> [811] 44.486707 31.296760 33.825723 29.673124 34.026282
#> [816] 165.011075 109.287485 105.432956 46.554563 31.546334
#> [821] 34.769495 26.309489 36.822224 170.078539 97.233150
#> [826] 103.437863 43.135479 28.860388 33.053009 23.208648
#> [831] 38.422893 159.336934 88.512512 103.206401 47.365989
#> [836] 31.572353 35.984752 22.722223 33.728057 146.322609
#> [841] 107.862552 105.438540 47.479481 28.481425 31.647929
#> [846] 26.164322 30.565289 149.787435 130.332740 98.296281
#> [851] 41.290008 38.151850 34.086264 29.415540 37.504760
#> [856] 201.926000 210.496647 93.751699 50.988839 51.721273
#> [861] 47.046052 29.129116 59.443360 349.692621 411.374657
#> [866] 85.913687 59.676289 90.024034 67.344532 40.821380
#> [871] 91.276277 530.047933 697.728942 79.454606 74.053664
#> [876] 132.664091 82.780161 49.933027 132.319325 649.397279
#> [881] 841.452977 91.543068 92.976836 168.231806 102.184256
#> [886] 58.848030 135.167703 733.247639 900.496888 94.106439
#> [891] 90.594612 144.141867 95.562042 51.211730 137.975992
#> [896] 645.368867 897.487709 86.226899 88.836395 139.545482
#> [901] 74.780693 50.911947 104.015507 608.068443 704.566139
#> [906] 94.131264 68.269341 92.880510 63.487855 35.247855
#> [911] 72.120525 399.302359 426.784746 87.379760 60.358757
#> [916] 63.452186 54.776193 29.311635 52.562169 282.731257
#> [921] 278.563776 96.429320 49.760013 48.450612 38.933191
#> [926] 25.844857 43.225975 212.039464 160.201024 98.777070
#> [931] 46.029153 37.862905 37.250173 27.008984 35.842104
#> [936] 169.997601 156.630668 93.014267 48.222163 35.064183
#> [941] 35.451472 23.543123 32.686649 171.065548 105.790041
#> [946] 108.830875 45.272460 31.981356 30.120026 23.585034
#> [951] 32.319106 159.294988 115.495441 115.896828 47.765108
#> [956] 29.215978 34.400212 25.209040 35.119547 165.818130
#> [961] 107.669547 106.891347 46.208358 32.254054 33.814447
#> [966] 24.801176 35.543628 155.108299 113.496329 110.971571
#> [971] 44.721446 28.139904 31.768555 25.935012 31.160421
#> [976] 141.595916 104.123449 109.959406 42.095975 29.355523
#> [981] 33.334306 28.015602 32.062996 150.583101 90.077886
#> [986] 105.341658 41.192123 29.251126 29.782968 26.674842
#> [991] 36.906096 170.581104 97.567593 105.959960 39.480080
#> [996] 28.286513 28.900908 22.996521 31.173936 142.981469
#> [1001] 93.127804 109.395854 44.881067 30.412322 33.138231
#> [1006] 24.754524 35.953917 140.121663 102.959301 105.862271
#> [1011] 40.174324 29.285178 32.617315 27.309924 38.589327
#> [1016] 166.073498 89.320133 106.740038 46.417630 29.564379
#> [1021] 39.726060 29.746300 34.125606 168.017869 90.316623
#> [1026] 107.312059 42.062266 29.621464 34.036371 26.995349
#> [1031] 34.642726 157.013142 97.627014 114.828925 46.800743
#> [1036] 27.051983 35.225135 28.283421 35.625325 175.540814
#> [1041] 118.424921 107.035100 43.272736 30.311741 34.189589
#> [1046] 27.176501 34.648530 180.816095 107.999549 106.611958
#> [1051] 44.284883 29.429024 34.550493 26.800240 36.931911
#> [1056] 156.397981 102.388683 114.543137 43.335283 29.670092
#> [1061] 36.463383 31.204052 36.964631 152.760545 105.690832
#> [1066] 112.532788 45.675366 31.084984 36.516981 27.889156
#> [1071] 39.951343 160.542475 108.768802 120.086017 44.621606
#> [1076] 29.979912 32.758499 29.640258 35.449074 144.627534
#> [1081] 95.904708 98.581966 29.151899 25.652984 34.015049
#> [1086] 23.739257 33.309044 132.845364 110.519425 97.435606
#> [1091] 36.445994 24.191037 33.755435 25.317860 34.692138
#> [1096] 133.370595 89.432865 78.788986 35.394559 32.745781
#> [1101] 34.401351 21.857643 37.652579 149.387315 92.966335
#> [1106] 117.034547 41.307684 25.799113 35.371046 30.828860
#> [1111] 36.504718 148.164662 173.861551 98.670328 50.817050
#> [1116] 24.953204 36.405526 20.948719 52.882848 151.105567
#> [1121] 391.764819 158.707810 32.949967 33.791382 46.733799
#> [1126] 21.774906 87.747553 160.094614 819.146131 141.687122
#> [1131] 41.526904 44.907794 62.191336 38.025100 108.899176
#> [1136] 242.205228 746.278612 165.371112 43.624310 52.703623
#> [1141] 95.556257 46.402372 204.718860 409.282334 1610.046856
#> [1146] 268.615342 60.597154 55.776300 92.681383 48.520625
#> [1151] 204.365289 371.091568 1585.529313 261.537745 35.539510
#> [1156] 62.214589 96.423575 29.725105 188.243389 314.450925
#> [1161] 1120.563718 255.135276 47.119980 48.516808 89.928485
#> [1166] 37.324552 142.475128 250.435000 566.727559 168.645876
#> [1171] 40.371340 44.392902 66.118259 29.763397 90.735531
#> [1176] 191.261879 789.235621 177.715166 49.942617 39.819652
#> [1181] 46.928773 29.820479 76.079930 172.221738 431.544954
#> [1186] 142.811706 44.438606 33.643979 39.060616 25.329606
#> [1191] 55.605822 151.479111 287.372645 130.826744 39.162669
#> [1196] 31.567921 36.137109 25.640328 36.565181 156.137129
#> [1201] 189.563114 114.160943 48.205198 27.381770 30.582241
#> [1206] 22.257580 40.508673 146.431932 116.312190 100.976158
#> [1211] 36.645808 27.605937 30.085477 26.043214 36.610432
#> [1216] 184.984871 126.509686 90.825038 36.832579 27.637315
#> [1221] 32.743402 25.398717 30.508300 140.194227 118.434799
#> [1226] 95.947858 29.674962 28.870998 33.375377 27.328116
#> [1231] 39.398779 178.195530 105.098561 84.987955 43.518142
#> [1236] 27.896133 36.033127 27.324011 34.533595 153.969597
#> [1241] 85.703834 99.892381 34.242776 26.875551 36.100405
#> [1246] 23.063659 30.791688 165.702633 67.073723 119.794516
#> [1251] 39.406710 22.804362 32.508115 31.149893 38.454433
#> [1256] 139.124003 99.513846 77.327728 43.546272 31.916123
#> [1261] 33.064523 31.806988 37.525104 183.940540 132.497918
#> [1266] 99.188331 47.054355 30.830371 34.161061 27.230252
#> [1271] 39.881592 186.165895 84.479401 96.370104 44.697988
#> [1276] 28.816349 31.995093 32.431530 39.123189 152.214855
#> [1281] 63.328493 124.861869 41.193206 28.866022 40.435678
#> [1286] 25.878933 35.254059 126.227156 110.655427 115.221361
#> [1291] 35.455676 30.530741 37.684927 17.316758 32.490885
#> [1296] 134.756237 105.090005 141.773908 34.771503 30.138122
#> [1301] 29.993710 19.134206 31.945019 174.943336 86.977711
#> [1306] 119.414188 46.054079 27.007063 37.394333 25.526424
#> [1311] 32.350547 176.360991 100.247268 92.214134 42.100218
#> [1316] 26.598997 31.552884 32.203519 33.252210 104.017945
#> [1321] 116.210076 62.106982 49.084530 26.358952 34.067227
#> [1326] 26.564477 36.290081 142.263964 47.620735 112.944846
#> [1331] 34.322501 28.551036 38.993419 22.362873 35.850753
#> [1336] 136.374939 99.798530 92.174216 24.876948 31.416041
#> [1341] 27.977401 22.599160 27.002678 136.757612 85.021712
#> [1346] 66.102933 48.928459 27.674189 31.963448 31.680869
#> [1351] 31.415532 206.067120 79.858983 67.724770 51.565559
#> [1356] 36.770206 33.478235 33.397290 30.300532 185.169355
#> [1361] 59.689057 113.624347 45.834682 35.071004 30.272516
#> [1366] 26.018613 31.311628 137.484421 97.771376 98.282888
#> [1371] 46.533382 32.927842 31.117646 27.572587 33.224553
#> [1376] 150.592315 72.236452 96.214203 52.651345 36.777134
#> [1381] 28.324229 32.304347 38.065710 116.620451 98.368924
#> [1386] 106.193656 54.976777 26.129837 31.190081 29.061710
#> [1391] 33.049347 235.458633 49.644926 105.635875 28.457881
#> [1396] 32.916288 41.376448 28.199523 32.091286 204.683036
#> [1401] 92.369982 117.488798 33.176475 37.188275 36.916034
#> [1406] 24.631105 35.289954 152.838308 74.815710 74.688163
#> [1411] 56.601092 27.957361 35.494486 34.825380 37.205302
#> [1416] 162.042605 49.309894 107.588334 42.188854 29.120570
#> [1421] 35.488853 19.608062 31.033799 186.844817 97.331014
#> [1426] 93.206562 48.051964 37.413884 27.915677 40.912424
#> [1431] 38.188606 119.108399 53.634031 105.031201 43.672971
#> [1436] 21.503384 31.892517 27.013946 45.004002 110.739233
#> [1441] 95.821644 93.103918 58.389068 28.267440 35.444377
#> [1446] 24.704700 36.539813 212.861125 103.871532 129.800170
#> [1451] 40.755682 30.372291 37.629196 28.002186 35.415052
#> [1456] 235.109370 80.404990 123.798571 36.751008 41.045761
#> [1461] 29.878926 24.239642 40.312472 127.568099 57.314662
#> [1466] 97.914116 46.504641 29.215859 38.111446 25.698333
#> [1471] 34.554269 157.990302 84.496738 74.541171 32.664953
#> [1476] 40.293004 28.729528 28.841443 38.652989 144.608685
#> [1481] 103.610632 91.145180 38.251578 32.525705 29.559268
#> [1486] 28.371520 34.002482 139.517177 47.311838 122.164686
#> [1491] 46.971462 28.612430 36.331883 27.220920 31.552084
#> [1496] 207.302292 91.903460 114.640190 30.747115 26.480455
#> [1501] 37.264549 30.834890 32.124568 183.139218 70.849133
#> [1506] 129.735962 40.666440 24.421463 35.638740 17.057095
#> [1511] 35.863203 160.275320 89.107102 123.293859 35.963508
#> [1516] 39.949520 35.584113 20.181869 31.828364 135.130744
#> [1521] 119.125820 96.000743 45.748627 25.493576 37.545546
#> [1526] 21.838315 35.173620 172.009139 70.488143 104.181722
#> [1531] 25.083607 32.690491 41.949165 15.853308 37.556713
#> [1536] 156.180366 89.693465 88.501997 23.182854 33.387140
#> [1541] 39.341711 19.210659 38.406144 160.599245 123.616225
#> [1546] 92.213588 36.325025 32.144818 46.369477 39.971955
#> [1551] 33.936432 195.647481 77.179203 111.316890 33.769247
#> [1556] 26.788419 46.454249 24.763817 39.588913 146.920034
#> [1561] 126.582540 109.726625 40.927068 38.069900 41.180900
#> [1566] 27.001062 34.850588 197.805155 80.765977 97.622657
#> [1571] 26.326894 27.408477 42.529164 21.699734 30.852826
#> [1576] 171.079778 93.319155 103.618405 31.096392 37.420658
#> [1581] 33.034041 21.859346 35.250289 171.183623 87.360047
#> [1586] 100.242504 32.113428 26.525980 42.798271 20.262626
#> [1591] 28.644451 176.150627 69.840818 119.224692 25.607179
#> [1596] 31.822188 35.727435 21.551686 35.178318 156.434354
#> [1601] 104.369524 101.711197 53.031272 29.573143 32.794880
#> [1606] 29.026631 36.193032 154.152449 89.099312 98.262950
#> [1611] 40.983253 24.163189 34.501748 31.976277 35.426385
#> [1616] 148.537631 97.701749 109.391669 46.714138 25.523892
#> [1621] 31.964336 35.362196 33.886820 157.480453 86.366383
#> [1626] 79.107788 38.764546 26.201341 25.624606 28.607790
#> [1631] 33.253877 142.411533 90.983309 93.451915 47.661561
#> [1636] 26.865360 32.096030 31.834129 31.258656 230.045142
#> [1641] 71.557235 132.132516 47.257443 24.495178 37.002452
#> [1646] 20.361930 36.194230 248.565752 64.068487 157.677227
#> [1651] 53.574143 24.249650 37.743398 21.712505 28.686528
#> [1656] 218.324965 66.007129 89.803788 46.140171 23.027989
#> [1661] 29.306930 27.006135 32.963365 187.819705 85.063270
#> [1666] 81.274325 43.366147 28.403436 37.554051 21.598555
#> [1671] 36.212499 158.448519 78.372012 77.477039 29.996444
#> [1676] 32.272852 32.906730 24.015367 33.172171 272.487378
#> [1681] 84.846888 85.014031 29.668608 39.581763 40.042868
#> [1686] 37.099377 36.948299 203.872117 61.969520 146.510894
#> [1691] 38.981498 30.018676 20.476881 36.929195 43.787444
#> [1696] 154.430081 90.605106 146.302234 40.669078 27.420887
#> [1701] 35.429644 57.348454 34.920677 263.241275 71.016427
#> [1706] 79.633341 50.394459 25.392463 40.220837 52.619259
#> [1711] 42.117045 215.196783 58.495471 108.490964 61.623339
#> [1716] 23.415285 38.055228 26.456648 61.001604 266.378869
#> [1721] 93.817655 121.125686 57.490407 37.148676 27.849703
#> [1726] 45.148767 50.306234 197.005061 106.544321 102.487807
#> [1731] 27.663152 38.055031 43.434172 48.045937 40.335960
#> [1736] 191.640031 67.029701 137.051483 65.579437 22.572407
#> [1741] 38.649261 40.508534 57.055165 140.799223 133.669713
#> [1746] 74.303656 56.998198 39.294406 36.815819 25.600391
#> [1751] 54.495908 295.211768 127.769307 109.169560 30.880279
#> [1756] 32.020910 43.418441 48.313316 48.187284 147.377280
#> [1761] 94.739802 119.769936 46.917040 39.250639 41.261021
#> [1766] 28.507046 41.979571 196.337320 81.106436 168.109136
#> [1771] 52.962343 21.686875 30.963254 43.027813 38.210871
#> [1776] 307.503439 80.468017 141.685174 47.534258 29.558209
#> [1781] 37.009343 37.449933 41.153272 166.665936 105.003535
#> [1786] 86.877174 55.994831 38.115102 45.375383 23.711647
#> [1791] 45.017327 195.095036 64.489254 141.102600 58.586026
#> [1796] 24.740041 39.417257 45.935621 44.368479 248.729436
#> [1801] 99.076422 108.528579 57.070673 37.177808 25.617253
#> [1806] 43.001979 56.238403 242.362830 68.406010 149.126453
#> [1811] 26.213064 36.171577 40.075221 32.053932 54.676368
#> [1816] 356.667826 55.901124 155.707960 67.176312 30.555825
#> [1821] 36.290777 51.125159 69.950411 172.199389 84.890150
#> [1826] 155.172876 20.486742 34.701371 45.691073 48.161121
#> [1831] 36.894187 302.110497 93.596723 91.851749 65.116098
#> [1836] 39.147736 36.466925 26.774093 69.335105 296.318700
#> [1841] 90.782109 215.716420 51.973785 27.257805 30.972503
#> [1846] 49.214708 58.873692 155.052388 131.714048 176.217341
#> [1851] 27.778185 31.060168 43.479056 37.612360 33.331283
#> [1856] 271.083706 108.032408 101.957330 52.877356 36.519604
#> [1861] 33.477509 23.041509 56.101345 305.169499 113.602917
#> [1866] 211.426262 53.749557 18.842347 39.586898 44.027395
#> [1871] 43.585771 185.544784 99.435757 98.333853 53.386409
#> [1876] 34.430626 30.946110 25.427338 38.509870 285.530040
#> [1881] 52.472315 198.993257 58.660556 38.832734 37.082364
#> [1886] 44.011392 41.031400 188.437410 87.474563 98.043931
#> [1891] 78.003673 34.698912 41.750663 35.420446 39.510279
#> [1896] 383.074116 98.765139 119.145008 51.552674 33.715571
#> [1901] 34.694111 42.824697 48.457643 296.863562 84.082733
#> [1906] 114.538239 28.738582 42.499469 43.984079 36.904957
#> [1911] 35.895894 325.381169 81.027964 137.467786 38.488739
#> [1916] 27.329626 47.504072 16.035063 58.288272 280.982411
#> [1921] 81.964843 147.251262 41.919116 28.917983 29.126941
#> [1926] 24.707881 54.181098 242.407159 100.310015 137.655731
#> [1931] 41.494815 26.177019 28.914556 26.650264 46.273476
#> [1936] 231.882754 89.895481 112.320566 32.803755 33.588569
#> [1941] 33.033767 26.608555 50.960213 458.659131 107.640478
#> [1946] 150.360576 65.517246 35.414668 38.215521 43.125685
#> [1951] 50.555097 283.978981 97.550146 135.993181 58.664513
#> [1956] 35.917116 36.995451 38.546234 49.043232 325.272207
#> [1961] 98.996154 147.221397 69.319522 35.468042 35.986838
#> [1966] 38.794882 50.329541 335.374756 96.065313 147.845948
#> [1971] 61.414690 35.989945 36.926313 38.591692 51.843903
#> [1976] 324.264422 101.529915 135.551127 65.209398 36.824668
#> [1981] 37.633576 39.007057 46.347952 316.556611 96.712077
#> [1986] 142.219890 66.475186 34.892562 35.069106 41.596664
#> [1991] 52.278679 254.675998 109.176872 154.631251 65.022175
#> [1996] 43.055877 43.341532 49.361935 54.482969 354.457471
#> [2001] 89.377830 160.112253 64.213553 42.558553 39.835207
#> [2006] 59.856009 60.354117 379.372871 109.208437 166.123293
#> [2011] 61.810196 39.636466 40.870663 51.759527 54.428493
#> [2016] 376.155093 103.506800 170.954710 64.387163 40.418625
#> [2021] 39.887188 55.752389 56.483176 362.650536 115.075388
#> [2026] 143.364995 57.492267 38.523954 32.647061 51.301592
#> [2031] 46.871748 384.224012 88.074090 163.403510 47.091308
#> [2036] 38.166879 36.352915 38.096904 53.610835 368.810280
#> [2041] 108.832158 153.158567 67.981493 44.443709 39.159903
#> [2046] 52.367430 53.014402 415.838201 115.424278 153.048996
#> [2051] 120.144144 40.145176 36.078019 44.489095 52.712724
#> [2056] 389.350239 110.070706 146.372008 163.313465 46.588510
#> [2061] 40.543530 47.885738 53.107820 399.845750 96.952821
#> [2066] 155.735393 141.124334 46.514724 44.535562 46.303457
#> [2071] 52.881468 407.918184 114.145907 158.973029 121.196010
#> [2076] 32.334025 36.807087 48.935408 55.988427 400.204529
#> [2081] 122.143079 148.332373 109.835709 40.129369 39.771837
#> [2086] 36.824987 59.225614 334.339159 105.224713 149.474562
#> [2091] 79.284124 46.528001 41.737876 45.739151 70.237961
#> [2096] 403.477595 117.842087 495.825624 74.954894 34.224355
#> [2101] 40.451839 46.355804 62.324136 286.454247 89.056594
#> [2106] 5832.395161 87.522507 51.302731 41.023700 29.757374
#> [2111] 48.602331 235.819508 67.292766 13873.384850 112.370406
#> [2116] 75.664919 37.635591 27.751099 37.371573 267.709799
#> [2121] 52.310421 17165.405181 191.168944 82.199662 32.221482
#> [2126] 24.518181 33.193361 252.173733 49.401413 16437.503614
#> [2131] 159.827322 89.323235 36.391296 22.471497 35.772829
#> [2136] 252.290946 65.733328 13755.012642 97.936771 60.497118
#> [2141] 33.123704 27.679177 34.807008 253.211050 56.774516
#> [2146] 10955.394431 96.865632 51.837906 29.911206 29.050536
#> [2151] 40.047838 234.478684 82.087474 9493.409165 83.231644
#> [2156] 51.672872 35.252748 29.158378 48.748037 365.643260
#> [2161] 90.750294 6442.331874 75.831233 52.951960 38.056488
#> [2166] 33.368845 48.303094 352.849221 91.706198 4600.186775
#> [2171] 69.476735 52.822776 38.611727 44.488193 51.911443
#> [2176] 431.401173 101.796074 2892.800990 66.987991 51.283043
#> [2181] 38.911728 45.438261 47.577080 399.170704 108.112526
#> [2186] 1774.720067 64.928496 46.699510 43.879691 40.686856
#> [2191] 50.936232 445.515064 106.386173 1354.003510 64.540338
#> [2196] 49.147460 35.080291 46.991590 48.374540 428.179441
#> [2201] 103.169871 941.571921 72.048556 53.248166 36.666954
#> [2206] 48.456305 46.269972 422.903522 110.674410 752.972866
#> [2211] 70.220963 49.831384 37.652212 45.846990 49.459702
#> [2216] 447.458198 106.774150 630.149374 69.259278 51.613374
#> [2221] 33.897051 46.393051 46.725414 449.146344 97.630969
#> [2226] 541.745739 75.886827 41.253986 40.926556 45.904882
#> [2231] 45.775102 470.359972 99.460572 494.868960 72.884706
#> [2236] 52.137230 42.584389 46.582839 49.957400 438.727083
#> [2241] 110.900816 455.857940 75.793757 50.861001 43.644965
#> [2246] 48.120407 47.968101 472.913455 113.232404 394.845602
#> [2251] 73.335230 53.254563 39.055537 43.107564 44.777950
#> [2256] 486.724239 112.924868 342.480613 71.166023 52.477110
#> [2261] 43.093652 48.508074 47.582490 473.236068 107.121591
#> [2266] 314.538700 68.552210 49.572075 46.860595 42.282366
#> [2271] 41.354109 435.768428 106.979632 279.561779 63.373882
#> [2276] 53.443520 39.888940 47.507710 46.408509 479.389012
#> [2281] 99.470195 295.953485 70.021186 46.548519 40.014569
#> [2286] 43.371476 40.823099 454.627431 116.416887 241.948283
#> [2291] 65.018561 48.924704 37.893611 42.659015 49.039957
#> [2296] 491.670790 95.426186 237.162205 68.319898 53.581855
#> [2301] 36.353723 44.225962 45.001981 447.547469 109.786524
#> [2306] 237.586439 66.047106 45.552718 36.644631 43.799834
#> [2311] 44.847455 418.299828 122.267204 222.628184 64.904554
#> [2316] 46.077932 36.239206 50.909309 45.872858 475.359280
#> [2321] 121.625129 213.582292 64.780518 46.055814 41.639253
#> [2326] 43.247804 47.021251 482.833683 143.198061 203.076683
#> [2331] 61.808888 44.536787 41.674611 50.834753 49.958632
#> [2336] 462.927389 133.091117 212.156884 65.674832 45.832769
#> [2341] 45.227610 50.943171 45.399300 476.837618 134.462510
#> [2346] 212.997852 83.683738 46.307428 44.114147 49.426245
#> [2351] 46.066511 445.476788 146.815803 205.989675 147.752024
#> [2356] 50.177558 52.419657 55.526217 47.264174 490.760576
#> [2361] 170.817562 214.155841 181.371703 57.437563 52.451408
#> [2366] 60.217505 50.999636 485.747137 160.952902 195.593397
#> [2371] 186.363268 62.290553 47.320707 57.157110 49.019309
#> [2376] 485.676005 143.149765 199.440368 135.085678 76.823418
#> [2381] 45.874357 46.694395 46.705219 472.472557 122.590519
#> [2386] 196.164505 114.501037 81.142977 42.737055 47.379126
#> [2391] 43.960697 469.841693 120.431267 179.920177 88.308295
#> [2396] 75.505423 40.908099 55.427963 50.543494 447.686020
#> [2401] 112.830110 183.489394 70.699660 60.818758 41.392392
#> [2406] 63.049013 48.161211 447.263685 113.098922 186.604226
#> [2411] 70.344673 56.597906 43.191166 63.650399 43.669881
#> [2416] 478.137230 111.280911 183.160843 68.099514 54.380999
#> [2421] 45.587965 63.487612 47.277363 435.605030 113.039216
#> [2426] 163.528049 64.942955 46.626150 40.701930 53.573868
#> [2431] 44.855228 428.627071 111.044178 183.114925 71.578556
#> [2436] 40.439535 44.389309 57.025407 46.306825 438.637845
#> [2441] 106.830294 180.154460 71.048972 44.054351 41.686204
#> [2446] 47.594233 50.640354 477.948657 107.768497 183.448420
#> [2451] 70.589348 41.367258 40.588047 57.552715 43.678015
#> [2456] 505.276160 106.934839 175.443230 64.234972 45.826434
#> [2461] 41.444938 51.237273 49.127882 456.103902 110.304658
#> [2466] 181.128785 64.361756 42.257462 36.234890 56.042026
#> [2471] 46.002784 461.831220 112.112989 177.799746 59.419955
#> [2476] 45.835058 37.143329 57.791545 46.574726 473.481790
#> [2481] 113.347008 187.479618 60.441204 44.384575 36.748130
#> [2486] 58.255789 50.525020 468.894201 112.097804 172.268805
#> [2491] 66.881132 47.767594 35.614725 62.727494 47.721740
#> [2496] 479.832255 115.216485 168.147780 64.495323 48.895200
#> [2501] 40.707435 62.435092 44.537999 477.044890 107.602994
#> [2506] 160.913590 67.899740 48.596475 37.600284 50.072798
#> [2511] 44.891404 446.804062 111.290585 162.397966 61.712172
#> [2516] 41.225893 35.227686 51.303568 45.314574 461.196746
#> [2521] 115.938279 177.651151 67.345495 46.733009 39.527198
#> [2526] 56.816790 52.750451 424.494314 111.871450 163.096121
#> [2531] 80.499247 49.625922 40.675166 50.999692 52.492706
#> [2536] 468.263938 113.744053 176.055005 122.768205 47.463385
#> [2541] 41.041710 53.205646 78.578701 584.468115 115.955268
#> [2546] 191.130047 181.661404 54.624808 37.018977 56.041601
#> [2551] 70.841264 664.795500 124.590019 224.475239 187.583307
#> [2556] 50.438938 40.283084 50.180244 86.135610 570.187038
#> [2561] 134.230020 219.730538 146.569392 50.101408 39.866188
#> [2566] 49.076670 72.141985 521.389886 117.411897 200.841469
#> [2571] 111.737539 50.180836 39.118886 49.177277 63.998615
#> [2576] 466.357814 130.208352 186.026854 90.567113 47.000837
#> [2581] 39.512403 47.244710 59.240777 451.402441 121.219202
#> [2586] 183.565928 85.706048 51.367771 41.658139 46.595095
#> [2591] 62.208513 461.008189 112.948330 208.372259 106.603505
#> [2596] 50.952128 40.728191 55.395050 68.092937 587.144867
#> [2601] 121.209110 245.040250 166.608778 50.820778 46.078316
#> [2606] 54.586626 73.634932 629.747347 106.906315 220.482740
#> [2611] 174.262815 48.301433 46.288178 55.616089 77.046168
#> [2616] 610.995708 103.686176 194.916945 156.926065 48.564475
#> [2621] 44.978622 52.375992 70.770942 566.716943 114.624499
#> [2626] 180.656253 109.721550 47.479018 45.371014 50.931045
#> [2631] 70.955909 522.202017 110.237149 173.128564 100.635447
#> [2636] 45.183336 41.858009 48.651565 64.857451 435.670086
#> [2641] 109.835674 173.932545 76.596251 49.585715 44.332319
#> [2646] 47.835264 54.787442 399.844011 105.368428 158.937927
#> [2651] 71.319149 45.394350 41.681597 52.281926 49.528685
#> [2656] 382.456539 105.509972 162.843921 77.230597 44.006242
#> [2661] 37.321572 83.274316 49.168353 432.168623 105.166536
#> [2666] 154.536989 63.360656 43.257950 38.976037 121.911508
#> [2671] 41.409602 416.626463 104.332561 150.248674 65.020749
#> [2676] 48.399111 35.372174 97.019543 41.577903 386.252188
#> [2681] 99.126223 136.614468 69.607545 41.745846 33.963000
#> [2686] 89.715885 47.772692 373.844217 108.473919 138.761989
#> [2691] 107.148913 45.289243 38.850900 66.693888 47.320003
#> [2696] 374.427710 100.337538 147.019071 108.946940 44.767658
#> [2701] 31.970107 55.714322 40.898479 384.554854 104.902908
#> [2706] 152.970046 102.968200 42.386481 35.469126 46.910892
#> [2711] 39.892412 392.099680 111.670846 158.119160 93.352925
#> [2716] 43.845470 34.770151 43.459377 45.528240 368.951429
#> [2721] 100.894014 141.036437 91.908482 42.468380 35.463312
#> [2726] 43.798070 53.840996 376.266252 99.509117 133.894132
#> [2731] 89.294837 39.262945 37.729908 43.829460 216.280428
#> [2736] 351.838721 101.478866 140.720373 82.380093 40.751900
#> [2741] 37.179003 47.645561 397.230075 362.989834 98.861373
#> [2746] 140.412077 70.692329 50.636567 35.765085 50.151279
#> [2751] 320.469480 356.958652 108.716669 141.060782 78.796086
#> [2756] 43.260207 37.810845 44.797668 278.136920 334.220326
#> [2761] 93.462171 133.870650 69.984913 42.031692 39.729968
#> [2766] 42.617221 203.946074 375.335101 92.918614 150.667486
#> [2771] 66.658469 64.518478 40.465141 37.987780 319.792309
#> [2776] 381.687629 95.203955 158.715536 62.906516 118.817601
#> [2781] 35.928716 44.179380 422.806755 339.826894 96.870755
#> [2786] 170.204776 65.174303 129.831309 39.115973 46.567989
#> [2791] 277.553212 379.195222 100.939231 160.881076 61.380293
#> [2796] 113.236567 30.495809 43.721927 221.316362 331.970851
#> [2801] 101.783902 156.075804 65.746408 94.056235 33.463657
#> [2806] 41.203415 134.397904 335.178189 109.343455 159.628160
#> [2811] 56.701886 75.474413 33.982118 40.438525 92.386664
#> [2816] 413.892579 107.377343 207.107832 58.226374 60.382087
#> [2821] 39.293829 41.553251 67.557693 502.811196 100.060179
#> [2826] 241.096155 63.202887 55.581887 32.281917 48.897676
#> [2831] 54.018214 530.671079 98.657496 250.577511 64.236887
#> [2836] 57.254045 29.696300 53.560764 47.510936 482.104900
#> [2841] 87.972094 243.426023 68.539853 80.170418 33.892214
#> [2846] 60.884106 52.454029 417.665537 95.028840 202.186937
#> [2851] 65.538071 96.851122 35.226911 49.554699 47.346519
#> [2856] 452.073756 105.329216 179.652847 63.027096 89.270784
#> [2861] 32.109190 50.222711 40.365956 390.430038 94.825371
#> [2866] 154.653021 65.312407 74.879131 39.632950 43.034229
#> [2871] 44.376038 364.625164 106.327953 155.579487 56.476849
#> [2876] 68.204701 37.557180 40.732334 51.897565 338.730040
#> [2881] 101.356362 152.716527 56.022610 64.241037 38.503367
#> [2886] 39.174198 48.377148 339.486165 101.688007 149.401876
#> [2891] 84.371173 66.150104 48.432286 44.200528 48.981838
#> [2896] 350.977215 101.010195 155.124837 261.774278 62.327187
#> [2901] 50.881668 41.499536 58.946579 426.405576 137.709243
#> [2906] 154.974981 1044.061565 75.624113 52.398541 56.741179
#> [2911] 90.532370 475.939693 236.967228 139.114540 3432.023477
#> [2916] 110.743161 75.961875 92.011387 166.027562 929.845177
#> [2921] 504.843100 139.855029 6483.536127 197.809790 140.472021
#> [2926] 127.368047 224.242597 1451.459637 717.187880 141.928024
#> [2931] 7367.100911 239.956124 158.085961 124.970249 220.324988
#> [2936] 1630.745826 600.373785 130.757386 6646.782293 195.933957
#> [2941] 165.239449 116.567387 190.198294 1142.097216 483.831937
#> [2946] 129.864168 5204.742301 162.181947 194.907652 100.592671
#> [2951] 155.622761 852.478428 309.304721 137.013250 4042.596232
#> [2956] 117.024751 183.219997 75.718323 108.972406 623.185056
#> [2961] 276.796656 138.015189 2280.942842 83.652375 146.110538
#> [2966] 57.217777 85.910595 426.909449 169.369923 130.765452
#> [2971] 1129.015567 72.541058 98.417314 51.786954 65.490581
#> [2976] 370.740360 137.832735 128.707394 584.099025 70.200104
#> [2981] 78.216922 45.082211 53.395062 396.197642 115.825544
#> [2986] 164.890511 344.759542 67.780839 58.186826 49.007957
#> [2991] 57.555344 377.432551 103.528653 173.131749 304.128630
#> [2996] 82.324333 59.280267 49.017062 58.471389 385.087567
#> [3001] 107.713102 188.159390 256.629299 76.506054 144.285021
#> [3006] 51.495279 51.635400 381.894585 115.585556 176.218209
#> [3011] 207.990364 64.513575 471.014842 48.795036 46.370679
#> [3016] 413.799603 114.708741 170.739805 142.628879 51.914241
#> [3021] 766.677505 46.983835 45.447159 385.264893 105.968021
#> [3026] 157.320836 124.018762 52.331639 727.865570 45.855819
#> [3031] 48.050871 464.501195 108.529287 140.250347 114.867799
#> [3036] 49.210129 1365.406863 50.159966 48.346549 401.379074
#> [3041] 94.303394 127.682224 76.557818 38.802320 8245.265938
#> [3046] 75.312788 41.553529 443.388435 82.071571 97.633059
#> [3051] 55.690245 35.804840 11817.493575 99.572008 41.501168
#> [3056] 446.000312 72.960672 84.298512 48.043065 35.754181
#> [3061] 12414.736519 99.395533 38.658161 397.013831 74.672682
#> [3066] 72.898924 43.723220 44.488826 12981.508815 108.049080
#> [3071] 39.232533 376.557258 79.987398 79.204333 40.242510
#> [3076] 44.137902 13369.054391 87.335052 34.362953 431.964433
#> [3081] 80.404728 81.348712 39.098851 42.970071 12251.031919
#> [3086] 83.383473 40.176316 405.543253 73.572794 92.162209
#> [3091] 47.164285 47.335381 11181.342307 80.073486 33.482190
#> [3096] 381.244893 82.093875 106.339604 55.248650 41.232214
#> [3101] 10035.242946 67.145501 41.769664 351.287240 79.788459
#> [3106] 116.131377 54.850998 41.936423 7552.693637 64.331488
#> [3111] 45.096367 363.235391 88.494106 152.857911 58.406151
#> [3116] 45.085574 5274.329443 54.984343 51.276605 384.683981
#> [3121] 85.207682 170.325455 56.245315 48.942358 3446.959797
#> [3126] 62.191727 72.638760 376.092814 96.718939 207.073727
#> [3131] 51.516924 58.128504 2306.458394 105.223533 90.857087
#> [3136] 437.576096 91.756143 202.965217 50.084695 171.950652
#> [3141] 1713.140865 201.747017 165.403270 574.701225 97.888899
#> [3146] 224.355435 50.426250 643.298990 1559.863603 357.425180
#> [3151] 250.534715 1457.781432 77.966053 327.988844 48.599136
#> [3156] 1830.193673 2340.699961 361.455326 221.523901 3259.837324
#> [3161] 80.292646 667.704245 46.440136 3960.048515 3318.063234
#> [3166] 374.118420 251.656618 4836.139255 75.492559 911.828175
#> [3171] 45.894324 5028.931315 3790.007761 376.525573 204.266086
#> [3176] 6447.558959 81.661543 1083.513819 48.934917 5462.590695
#> [3181] 4040.078162 316.379302 201.778532 5451.933901 77.594644
#> [3186] 1029.314338 50.048455 4427.503115 4019.770812 354.163547
#> [3191] 159.297537 4770.406330 89.531190 681.997054 50.058454
#> [3196] 3735.746069 3014.155752 253.142876 138.573892 3442.385517
#> [3201] 108.520769 567.223315 48.193977 2389.961043 2112.600046
#> [3206] 167.179547 116.898048 2200.591211 90.379031 389.386558
#> [3211] 58.841952 1387.697525 1671.304308 122.009446 91.749983
#> [3216] 1334.821787 86.827529 273.706301 47.739604 682.037909
#> [3221] 2702.578604 116.541629 68.538327 1053.298147 90.165921
#> [3226] 199.887662 48.969506 366.617997 8110.564172 119.166472
#> [3231] 57.492952 1253.169747 79.696759 125.325973 70.386734
#> [3236] 185.103845 17817.324496 154.758688 54.444984 1848.090470
#> [3241] 86.306052 112.925436 69.415037 97.107928 24421.982208
#> [3246] 177.387707 53.053684 2350.667822 68.231133 115.283247
#> [3251] 70.470103 67.408725 24224.176889 199.367430 54.483140
#> [3256] 1851.521975 71.252109 104.026548 73.399323 62.840590
#> [3261] 20173.169426 159.624412 54.335656 1643.522781 75.692504
#> [3266] 112.373523 84.076986 71.224743 15413.772897 94.253645
#> [3271] 46.128246 1353.106928 87.476977 123.569037 76.496362
#> [3276] 83.747586 10410.026459 73.003667 43.600309 914.755517
#> [3281] 86.775649 147.780305 77.343147 80.636035 5586.789067
#> [3286] 59.589081 41.941584 542.935288 94.678388 156.837136
#> [3291] 111.355258 84.365073 2892.251637 49.823283 51.304918
#> [3296] 529.727005 106.611109 202.748799 118.293521 114.514784
#> [3301] 1277.830641 57.576321 46.114399 422.564773 105.676005
#> [3306] 216.152271 131.173524 98.990215 730.104704 56.522926
#> [3311] 53.390978 384.438329 93.755092 285.822187 97.594344
#> [3316] 85.205392 547.206071 46.915710 51.918508 404.797401
#> [3321] 93.750448 295.711001 140.678887 77.257286 421.634536
#> [3326] 46.707930 54.991718 377.567921 100.374922 312.936990
#> [3331] 132.502633 71.580301 274.483526 49.194447 61.477936
#> [3336] 431.160271 88.113807 354.666093 124.343121 70.724463
#> [3341] 237.235026 53.864975 57.278502 386.820263 99.928820
#> [3346] 387.976732 135.955634 88.630018 214.309572 54.529801
#> [3351] 54.718577 337.031514 114.828256 287.822821 156.152333
#> [3356] 119.974355 180.442025 69.066942 51.631361 386.171758
#> [3361] 110.462710 307.941976 156.382602 142.630032 144.549083
#> [3366] 53.404418 50.098636 364.021570 140.529983 246.337782
#> [3371] 215.574043 150.186601 143.793425 44.319647 49.858377
#> [3376] 425.172901 146.985848 220.863097 398.886785 190.947449
#> [3381] 121.991410 53.573488 47.726592 383.298964 129.257191
#> [3386] 217.825355 747.478983 243.704667 125.857035 54.983686
#> [3391] 50.734335 448.644560 128.739227 227.458562 1514.914099
#> [3396] 446.209846 125.520585 50.398286 59.134788 446.003391
#> [3401] 107.173052 231.371720 2620.309723 705.822113 115.209906
#> [3406] 53.223066 70.486035 459.659304 106.569502 337.338604
#> [3411] 4899.363778 1153.960002 134.494480 56.529112 78.164909
#> [3416] 583.802260 104.608224 499.497467 7395.244639 1887.052951
#> [3421] 211.215561 94.945173 103.651928 788.532002 106.056672
#> [3426] 800.572374 10043.866250 2623.693823 240.230819 111.280702
#> [3431] 148.987083 914.847067 109.614644 1249.143255 12097.695780
#> [3436] 3653.450436 277.743610 182.055874 179.234241 1221.271458
#> [3441] 126.401723 1715.142546 14563.587860 5407.566131 264.512774
#> [3446] 210.784998 229.734521 1587.126612 124.967387 2232.878777
#> [3451] 15532.657079 6234.608575 269.324034 208.424586 233.263276
#> [3456] 1552.763248 120.786471 1960.332508 15743.664269 5880.225753
#> [3461] 220.480492 230.471743 231.182513 1374.267268 122.930560
#> [3466] 1785.462605 13040.903736 4625.013483 185.892917 177.027535
#> [3471] 207.044462 1064.031657 129.958451 1151.740920 10275.138221
#> [3476] 3030.566003 134.904623 101.688033 129.392091 737.169609
#> [3481] 110.267769 682.744596 7212.835770 1644.275912 96.058334
#> [3486] 74.493444 88.870497 520.998709 98.802609 438.159170
#> [3491] 3896.246661 903.259424 80.395037 70.742299 64.676344
#> [3496] 463.621414 92.772626 298.435963 2059.432492 466.110953
#> [3501] 74.424627 65.230299 55.563819 424.500233 95.895749
#> [3506] 218.235428 1111.334132 276.446303 70.360008 58.971164
#> [3511] 51.898553 421.426599 94.400382 183.128169 641.387997
#> [3516] 186.855560 64.828225 61.017041 46.322119 405.869448
#> [3521] 96.276210 165.488380 444.988146 134.354429 68.978034
#> [3526] 59.648629 41.928908 390.248778 97.680430 142.669647
#> [3531] 326.401134 100.406484 69.730199 56.872277 47.295864
#> [3536] 410.286925 100.654992 147.297034 253.873683 88.259557
#> [3541] 69.272909 54.819753 53.691147 357.119705 96.424177
#> [3546] 159.983678 197.203818 70.524063 64.887608 59.124792
#> [3551] 55.327900 392.013416 112.728092 166.700046 165.703903
#> [3556] 70.800969 71.877032 53.680604 150.170933 386.079010
#> [3561] 128.308721 142.192752 116.723960 63.349980 94.577026
#> [3566] 52.785717 618.750584 350.651580 115.429550 162.722968
#> [3571] 86.215457 51.653908 203.842317 53.626194 2246.121891
#> [3576] 382.415158 102.911678 134.274657 65.262824 67.883797
#> [3581] 529.226182 68.566852 6140.326677 470.275514 86.503963
#> [3586] 120.076985 52.290525 95.471138 1327.676572 152.403422
#> [3591] 12409.546012 446.365777 81.111320 120.051437 38.316607
#> [3596] 125.396044 2111.053553 192.086990 15265.605845 505.665088
#> [3601] 68.267354 110.300507 42.960764 122.990922 2434.511379
#> [3606] 193.448085 13856.641640 473.746327 61.488787 118.927335
#> [3611] 40.846413 98.481253 2635.015115 145.921496 10782.008108
#> [3616] 514.134987 75.622974 108.849099 43.482325 72.364911
#> [3621] 3990.963925 135.316360 7795.630482 619.633191 76.814228
#> [3626] 96.248114 42.018788 61.545582 5406.427674 89.845524
#> [3631] 4769.421076 970.879131 75.477365 85.024211 40.636458
#> [3636] 56.770813 5599.366446 84.311730 2351.550069 1181.581148
#> [3641] 78.535752 87.937028 51.470706 80.762274 5635.438907
#> [3646] 160.984848 1184.728514 1395.521664 100.176642 86.781772
#> [3651] 73.051767 160.614398 5912.221951 219.380200 738.510947
#> [3656] 1140.732565 145.020645 84.483414 86.219270 289.808744
#> [3661] 4809.441537 317.619402 519.229572 955.273766 138.266457
#> [3666] 86.975240 94.872176 315.805714 4244.719148 287.564623
#> [3671] 385.001572 745.416471 139.109839 94.263892 89.964926
#> [3676] 245.282922 3344.973796 379.042695 285.002069 562.866452
#> [3681] 118.547819 93.237187 90.176100 196.391278 2100.318811
#> [3686] 372.299956 239.873667 524.349564 106.931143 106.563291
#> [3691] 76.841937 139.732984 1207.312222 316.286884 197.681512
#> [3696] 436.388388 91.078274 113.614040 71.921476 96.063303
#> [3701] 652.662973 256.810664 168.067726 441.319650 88.802723
#> [3706] 109.248382 81.351967 73.164431 396.452972 225.406458
#> [3711] 142.344921 374.552271 93.302201 111.343348 67.648089
#> [3716] 70.830980 300.911453 204.240726 133.175767 342.959788
#> [3721] 87.580712 113.913341 69.461376 65.748910 411.875001
#> [3726] 308.109618 129.885858 418.053823 88.435850 126.543285
#> [3731] 58.067230 82.053081 1163.347622 656.206055 142.131827
#> [3736] 483.019800 81.782910 125.925370 55.778440 134.806233
#> [3741] 4152.926938 1369.659515 175.497921 1422.080153 74.546009
#> [3746] 142.809977 57.799691 158.359331 10781.055266 1887.565770
#> [3751] 254.454628 4125.279513 57.171463 159.062548 49.352282
#> [3756] 191.586048 19414.716845 1915.301956 249.685607 9593.457980
#> [3761] 53.229879 179.321117 49.398018 219.538918 25027.612744
#> [3766] 1699.348942 237.978199 12125.899330 47.666146 150.118401
#> [3771] 45.483353 205.384385 24722.054114 1560.084209 187.063079
#> [3776] 10025.053218 47.770314 142.855425 45.881075 160.115075
#> [3781] 22247.285425 1122.209056 179.858794 7811.386646 57.296257
#> [3786] 139.605985 42.877661 120.245360 17227.041343 966.745916
#> [3791] 160.845884 4808.898843 69.792791 125.729821 38.167593
#> [3796] 84.820290 11515.139382 548.766364 115.779634 2597.306317
#> [3801] 74.623623 115.060676 45.678531 75.170218 7086.094049
#> [3806] 378.804598 96.321522 1232.934934 81.323214 120.188202
#> [3811] 56.723953 66.465334 3185.372583 268.484301 82.963581
#> [3816] 687.802587 85.506730 117.224576 64.479412 65.524019
#> [3821] 1814.608266 225.544707 85.407348 542.782638 86.349111
#> [3826] 123.004558 58.317664 66.202576 1100.415739 214.043717
#> [3831] 77.217544 388.616710 81.948864 124.003550 70.615695
#> [3836] 61.841084 667.588209 172.941852 72.987441 404.906494
#> [3841] 91.581221 116.061535 69.550928 61.093073 432.550454
#> [3846] 190.066188 68.221521 423.191811 86.533175 132.812863
#> [3851] 63.531707 67.118890 403.301821 192.687840 73.299575
#> [3856] 444.097988 89.506922 113.664797 60.807217 65.998789
#> [3861] 407.663872 164.538277 124.435118 553.638583 86.238665
#> [3866] 130.324484 70.253929 61.002073 435.633906 152.830988
#> [3871] 391.973385 547.802014 81.861402 112.163967 59.381364
#> [3876] 59.971323 411.557948 135.541161 1744.958889 612.703115
#> [3881] 88.034656 110.804463 61.708663 71.230467 293.507325
#> [3886] 110.757471 6077.740919 862.483115 81.360095 105.068825
#> [3891] 67.573796 81.850953 186.911564 111.805663 13307.111044
#> [3896] 1372.664704 70.698866 88.051398 82.856088 108.482019
#> [3901] 144.194354 85.800517 18869.675912 1635.211647 64.893472
#> [3906] 88.788734 104.189162 113.902982 109.419341 84.412407
#> [3911] 20187.201067 1944.522540 63.006302 102.301590 93.458797
#> [3916] 111.017181 99.386746 89.749888 18506.676232 1730.000063
#> [3921] 62.605072 139.625281 84.196161 92.953162 83.759750
#> [3926] 76.876001 14237.193458 1203.023799 84.155309 189.537974
#> [3931] 77.877443 70.071738 80.401970 103.007223 8360.494762
#> [3936] 828.841696 84.691190 241.267317 65.904531 64.646327
#> [3941] 83.996235 113.440815 4721.723304 635.543387 90.308433
#> [3946] 222.556461 53.405444 55.582060 78.115123 125.319860
#> [3951] 2118.763134 487.729132 93.171913 192.335782 61.750752
#> [3956] 56.489347 108.363090 132.220562 932.305071 423.365816
#> [3961] 91.165989 145.139294 80.454267 51.459135 306.211543
#> [3966] 72.197537 439.134534 393.756957 76.900078 172.189370
#> [3971] 112.221617 34.701941 937.758675 87.240965 236.845782
#> [3976] 579.096423 102.674654 293.163169 327.418643 34.228399
#> [3981] 3036.340119 137.194945 179.647357 1120.911737 109.453593
#> [3986] 971.508357 1178.908284 39.262406 7097.736498 177.924146
#> [3991] 192.667422 2053.846316 278.958163 1660.497186 1904.867305
#> [3996] 47.415967 13612.566461 342.179352 294.165621 3160.448690
#> [4001] 435.425394 2127.446369 2887.688182 42.565342 14766.058057
#> [4006] 345.222552 278.494640 3322.225743 372.767125 2362.250764
#> [4011] 2609.591924 38.852476 12625.212351 309.212933 222.647444
#> [4016] 2664.950264 315.022733 1834.223903 1730.079802 38.026996
#> [4021] 9392.282426 224.407183 190.014574 1941.780229 207.846138
#> [4026] 1009.193277 1040.100975 40.215923 5706.949405 163.142562
#> [4031] 250.473155 1476.288686 148.233481 633.366188 545.469896
#> [4036] 40.898872 3206.350817 153.621670 1082.551877 1200.967449
#> [4041] 119.793940 425.444598 296.827609 42.566649 1538.463505
#> [4046] 268.504453 3411.768063 1179.528350 117.978419 465.694062
#> [4051] 182.231784 59.051107 707.841693 798.219025 5707.023102
#> [4056] 1486.346857 117.639523 831.651246 192.140575 69.683965
#> [4061] 487.604232 1810.516731 5655.658358 1759.984767 138.435596
#> [4066] 908.943295 205.512557 62.792249 586.376364 3350.792698
#> [4071] 3557.345636 2794.186745 96.937053 1355.829542 229.861769
#> [4076] 122.015590 1829.677277 6027.546728 3410.287913 2622.834168
#> [4081] 77.087682 1324.298012 191.503052 163.245090 3664.284597
#> [4086] 5437.512162 1735.487584 2429.998068 108.438155 1178.696880
#> [4091] 162.410241 267.114850 6283.259947 4333.732537 1024.766212
#> [4096] 1805.691424 108.535899 706.189219 91.852790 244.940871
#> [4101] 8028.558449 2034.124100 643.886912 1045.987358 68.133528
#> [4106] 330.332173 52.346737 154.506984 8406.522748 1084.783013
#> [4111] 396.354323 536.963676 54.900689 156.037455 36.911622
#> [4116] 100.574239 8086.313752 465.948604 289.557508 323.240735
#> [4121] 51.519895 121.549670 37.724636 70.075937 7054.943526
#> [4126] 284.338271 164.873854 264.384663 44.183074 94.368913
#> [4131] 33.475381 53.233161 5233.526518 206.557768 148.569036
#> [4136] 229.047964 47.795181 101.349570 35.298529 43.934168
#> [4141] 3946.345265 164.245997 129.941261 229.565143 52.575088
#> [4146] 81.682126 40.134699 42.011162 2149.719056 180.495567
#> [4151] 153.656255 252.291856 51.914673 96.644777 39.215569
#> [4156] 44.699284 1105.301500 170.612759 191.196159 299.601564
#> [4161] 55.762865 89.791462 37.016342 48.470076 631.509387
#> [4166] 142.981072 313.874315 261.312555 54.545496 96.572753
#> [4171] 38.547161 40.580551 403.145616 144.833464 342.246587
#> [4176] 310.954171 64.032320 98.969568 41.323106 47.257789
#> [4181] 320.515738 136.787292 286.709227 266.342598 53.949720
#> [4186] 100.135118 41.356455 43.328885 267.502695 144.459614
#> [4191] 164.988137 328.513179 57.461130 94.882088 40.005957
#> [4196] 39.564587 207.814849 126.104782 119.881971 280.057880
#> [4201] 52.793556 89.049451 35.078434 46.238785 146.926702
#> [4206] 110.740945 84.288822 279.134654 47.290907 94.405094
#> [4211] 37.226860 48.481465 162.433349 106.836158 77.830367
#> [4216] 264.453547 64.449780 88.148473 51.289629 40.248770
#> [4221] 127.812709 104.602114 67.768942 258.024133 61.969323
#> [4226] 82.904072 41.338179 42.952148 110.149472 117.332622
#> [4231] 67.180968 204.606299 56.525766 90.216799 46.333286
#> [4236] 44.435855 99.924897 100.880076 64.386607 258.448507
#> [4241] 66.325437 93.075675 36.416222 40.134242 95.424911
#> [4246] 120.528329 62.859443 261.412888 61.412603 86.133227
#> [4251] 42.906540 39.572776 82.769467 111.134468 51.519102
#> [4256] 286.668917 64.283166 84.813246 40.286968 37.904904
#> [4261] 69.903974 101.585087 56.501014 278.245946 61.309376
#> [4266] 87.758078 39.301138 41.492513 65.751722 104.708166
#> [4271] 52.745788 289.672285 62.170847 84.853457 36.613074
#> [4276] 41.065433 63.258503 105.844881 55.619390 279.935325
#> [4281] 65.660522 80.181659 38.893459 40.557632 57.631244
#> [4286] 128.399536 52.285071 293.503915 56.622564 95.275258
#> [4291] 45.338733 45.787694 54.856984 98.758217 53.656562
#> [4296] 293.183126 46.206202 110.215465 74.328091 56.813193
#> [4301] 66.842069 118.049178 58.332928 357.771687 56.772910
#> [4306] 123.815829 89.686368 71.647151 56.830797 153.497084
#> [4311] 49.366267 331.818114 56.743310 116.216086 76.397035
#> [4316] 59.559612 59.977540 145.390264 49.855219 314.519660
#> [4321] 61.537938 121.968848 67.554024 61.811850 57.497588
#> [4326] 129.414189 49.220161 280.976827 58.198151 109.382159
#> [4331] 58.828400 63.334645 51.710396 158.309001 53.081195
#> [4336] 233.696873 54.915254 99.290802 37.056606 53.601216
#> [4341] 50.327137 161.450047 65.423462 192.412946 50.879132
#> [4346] 92.203203 51.482459 50.083648 51.578465 137.690177
#> [4351] 82.735760 282.085141 49.772270 92.265072 68.277279
#> [4356] 73.567708 58.589771 142.897967 86.865603 321.770474
#> [4361] 56.413381 206.576746 171.211971 131.399072 46.079827
#> [4366] 159.835466 177.649695 363.398232 56.437854 584.305911
#> [4371] 571.609175 323.764918 46.295060 228.783927 426.355290
#> [4376] 542.757191 46.243221 1658.880072 1622.507155 997.029544
#> [4381] 52.807188 531.293593 749.134423 922.991166 39.452485
#> [4386] 2825.488790 2877.784777 1508.763285 65.139674 831.675763
#> [4391] 1141.031608 1391.305612 39.270631 3856.434958 3860.638527
#> [4396] 2222.188867 71.057165 1332.707846 1264.313461 1480.532561
#> [4401] 40.535523 3580.879003 3477.841466 2148.575595 77.281534
#> [4406] 1014.496104 1115.037402 1125.428625 39.748277 2954.830371
#> [4411] 2742.905155 1478.568493 61.673042 600.030137 762.467306
#> [4416] 751.502703 42.944433 2058.174511 1747.792682 818.145429
#> [4421] 47.708205 379.688054 512.696804 591.761472 42.598034
#> [4426] 1085.150648 920.500510 370.857232 43.804657 257.972650
#> [4431] 373.951200 475.887365 55.008088 646.133952 497.478089
#> [4436] 222.843946 41.230330 194.674755 178.710725 371.505328
#> [4441] 57.140406 339.753634 231.120987 134.328742 42.509141
#> [4446] 153.446260 132.452704 337.295803 54.562566 238.367276
#> [4451] 181.902731 99.626090 45.039130 152.466819 123.765795
#> [4456] 319.224321 53.479184 191.147905 133.862770 77.500712
#> [4461] 40.884697 144.562822 89.399850 310.357274 50.228462
#> [4466] 148.823911 97.549715 72.132407 39.522509 130.002939
#> [4471] 77.423112 289.631985 55.407644 142.645613 103.648934
#> [4476] 71.219004 51.694390 139.958523 82.921971 292.494079
#> [4481] 52.205973 181.637192 69.654248 81.094329 68.157062
#> [4486] 169.031478 86.042279 308.772410 53.377927 310.139904
#> [4491] 69.020225 66.398735 76.538431 163.269922 74.792746
#> [4496] 264.083464 48.114779 829.376209 69.830268 66.752727
#> [4501] 76.916664 163.950370 86.172492 276.670364 52.656940
#> [4506] 1914.579471 65.068199 58.827195 81.856336 196.329029
#> [4511] 104.180491 502.488765 53.568452 2747.098598 51.924475
#> [4516] 59.908836 87.904676 202.476039 125.925033 1019.848396
#> [4521] 53.380118 3511.184929 48.108025 56.457501 150.473284
#> [4526] 227.412918 142.088304 2872.239379 56.085864 3064.459565
#> [4531] 70.128100 63.608724 241.978705 214.168285 127.073370
#> [4536] 7327.306561 47.353124 2551.168664 69.940965 78.336143
#> [4541] 241.413151 249.864527 163.291762 11816.092896 44.269472
#> [4546] 1506.343634 55.429840 75.701900 178.393479 213.936204
#> [4551] 149.746016 10828.569943 51.917788 875.938513 50.516516
#> [4556] 91.879225 128.072299 243.998080 124.920143 8187.996344
#> [4561] 52.379129 452.274888 42.016104 67.711327 86.443328
#> [4566] 215.559648 101.002137 4219.553405 51.288061 276.236507
#> [4571] 38.036253 62.686607 70.009414 187.261870 91.053534
#> [4576] 1847.232543 55.696516 199.953871 36.243559 58.281916
#> [4581] 57.880514 191.821269 82.974095 941.544817 50.940288
#> [4586] 163.811216 38.687191 51.246496 49.906409 169.517866
#> [4591] 73.059557 643.602890 55.092397 133.211018 39.873766
#> [4596] 49.214188 48.038795 168.921205 67.181868 493.367367
#> [4601] 50.925881 118.515011 31.855963 52.775020 46.840387
#> [4606] 179.739927 69.377023 411.059316 50.439892 112.860340
#> [4611] 32.885993 50.016701 48.796571 166.045830 63.744490
#> [4616] 379.992069 49.849600 125.535197 36.394381 42.845141
#> [4621] 44.649761 183.210385 67.217464 317.219016 55.596707
#> [4626] 92.889999 34.362905 50.603551 48.219711 200.643150
#> [4631] 58.318596 312.149223 49.672869 92.801931 31.449582
#> [4636] 48.793051 45.627728 183.778058 52.802457 302.197141
#> [4641] 51.193953 80.921134 39.615357 46.794926 47.705001
#> [4646] 180.875624 52.438282 276.114152 50.887574 82.552679
#> [4651] 36.917704 47.417947 40.085574 175.438082 61.262524
#> [4656] 286.949399 53.042678 103.432709 33.243855 53.685708
#> [4661] 49.722538 185.507442 65.283741 287.849976 52.636642
#> [4666] 84.107162 31.474252 63.818638 48.940789 164.824659
#> [4671] 70.795252 264.853286 59.222573 90.152780 27.160904
#> [4676] 89.749870 57.063146 181.639975 66.890297 269.435757
#> [4681] 66.396097 80.147617 28.577496 122.774279 71.885516
#> [4686] 178.217865 59.325944 247.322788 68.980612 85.526426
#> [4691] 30.771644 111.374158 85.778464 230.254863 59.405668
#> [4696] 262.859357 56.453945 80.841921 25.095487 111.384500
#> [4701] 101.615747 292.653718 75.714277 236.219201 62.770011
#> [4706] 76.347353 29.549898 92.813874 94.254533 277.517573
#> [4711] 53.902052 232.997644 59.397059 71.790755 29.417685
#> [4716] 80.426822 88.550111 244.533126 56.060521 237.362021
#> [4721] 55.113275 72.179748 30.059435 68.114589 64.858636
#> [4726] 246.275348 48.147082 231.276261 59.358420 71.755033
#> [4731] 31.014290 61.453193 56.600685 207.804537 44.582336
#> [4736] 216.942810 58.470191 74.902481 29.037620 56.128376
#> [4741] 41.433763 214.985116 47.945246 238.681643 54.878736
#> [4746] 74.306264 28.871119 53.933283 51.477292 240.338017
#> [4751] 45.071880 319.892479 60.243923 71.908718 30.792017
#> [4756] 50.461645 43.276440 228.400380 49.487292 296.458126
#> [4761] 46.205151 72.394365 31.631879 51.444877 44.081211
#> [4766] 264.518612 43.777099 331.151428 52.130969 75.112764
#> [4771] 34.393221 46.578332 42.388637 276.102014 56.115048
#> [4776] 362.837851 58.398589 74.458122 30.720373 45.265009
#> [4781] 44.877775 276.971183 52.895040 349.033458 46.847567
#> [4786] 72.428151 34.847921 63.150338 52.859219 278.265463
#> [4791] 53.059548 396.797879 47.275672 66.330887 29.480862
#> [4796] 71.637194 53.837807 307.917395 56.975557 497.815422
#> [4801] 48.149000 72.074457 30.924934 93.585150 44.845495
#> [4806] 333.082104 64.602899 628.595244 46.241044 68.278919
#> [4811] 40.911953 91.214876 49.440920 293.720754 60.020386
#> [4816] 627.625701 50.614990 69.169435 28.467986 70.115238
#> [4821] 43.505653 309.938003 59.928173 575.279742 47.659137
#> [4826] 71.464793 32.106199 60.286711 49.790274 294.248272
#> [4831] 58.653218 685.388791 46.213836 69.006738 29.242110
#> [4836] 51.410295 45.520157 357.318091 51.821616 703.832977
#> [4841] 47.358509 75.602755 28.371720 51.350037 48.103827
#> [4846] 420.375025 54.557280 660.463500 55.045969 76.902699
#> [4851] 32.517357 51.046595 47.326629 455.927849 46.463520
#> [4856] 652.636737 49.820302 67.005941 32.962791 56.354765
#> [4861] 39.792577 535.631324 50.786102 482.542554 45.937160
#> [4866] 66.954484 27.526334 57.859302 47.467405 676.631754
#> [4871] 47.187554 475.886387 45.488405 68.867610 28.043411
#> [4876] 66.464158 53.735365 990.586320 48.262246 486.732631
#> [4881] 47.613015 64.115459 26.575104 89.239618 78.133335
#> [4886] 2147.019938 60.448976 485.854286 46.575728 66.852067
#> [4891] 25.055291 140.143325 119.297522 4785.156591 79.674124
#> [4896] 468.269479 40.069633 53.752282 21.424326 230.675430
#> [4901] 179.378576 6983.051321 108.451029 604.748155 36.021672
#> [4906] 56.476180 25.989365 277.886080 238.432355 8121.600158
#> [4911] 113.564386 529.230850 34.702434 56.259035 24.925178
#> [4916] 263.869149 208.314547 6791.957569 115.353383 462.317766
#> [4921] 42.774459 62.295376 25.392242 182.715289 113.173815
#> [4926] 4407.928285 90.005843 380.251795 46.900266 55.914632
#> [4931] 27.799285 115.570539 79.058044 2517.681085 67.723625
#> [4936] 365.169886 46.346011 63.736670 26.285887 63.575800
#> [4941] 54.973395 1016.756863 59.035719 314.809195 42.061985
#> [4946] 57.064270 24.355705 49.483228 41.642904 629.549182
#> [4951] 46.952355 299.548207 43.353903 61.252663 28.244145
#> [4956] 44.116027 40.359105 413.014250 42.694832 348.094255
#> [4961] 44.406247 60.548911 23.920501 41.520794 37.626969
#> [4966] 280.745009 44.364923 276.202102 49.087521 69.952603
#> [4971] 29.107688 37.645823 37.956093 187.936462 49.330026
#> [4976] 271.683895 49.996027 72.130079 24.524474 36.834286
#> [4981] 34.680823 173.507615 48.710235 308.370965 49.999272
#> [4986] 65.250505 27.344688 37.068941 29.006739 126.356819
#> [4991] 53.180806 325.906607 45.791061 65.085692 30.906553
#> [4996] 42.035999 33.820820 115.196643 48.461630 282.270670
#> [5001] 47.862877 65.823179 27.003224 36.460184 31.905152
#> [5006] 86.673064 53.963977 334.402929 45.000086 68.865902
#> [5011] 26.074596 33.596923 34.060453 76.903663 56.431894
#> [5016] 328.876977 45.333796 61.137680 25.360921 34.779504
#> [5021] 35.907375 71.257939 54.631313 321.782398 55.947935
#> [5026] 75.419724 26.057713 33.953019 33.511435 62.095741
#> [5031] 48.594550 318.331998 50.517598 62.658040 26.447861
#> [5036] 35.184809 35.683931 49.447750 45.818880 297.853359
#> [5041] 43.528390 64.768476 25.413464 30.008566 33.274146
#> [5046] 54.379836 43.844371 277.684504 47.210207 61.803186
#> [5051] 28.459141 36.008184 32.183616 45.571559 43.043387
#> [5056] 216.424570 47.089564 61.269947 26.100267 31.252960
#> [5061] 30.096715 43.530337 63.426359 240.832936 45.444751
#> [5066] 69.823334 23.452219 34.694233 28.758334 43.586328
#> [5071] 228.805007 271.242768 51.764197 59.215860 25.704444
#> [5076] 35.203498 33.849074 57.042047 883.215340 198.589383
#> [5081] 46.525925 59.050193 25.690238 33.508681 34.143498
#> [5086] 103.191358 3793.034272 237.970979 46.374508 63.158940
#> [5091] 23.593530 34.508682 37.986472 321.577456 10387.647943
#> [5096] 221.502027 46.100283 40.960708 20.854672 40.466064
#> [5101] 48.014631 458.881079 14625.467060 334.037362 33.822183
#> [5106] 49.832747 22.347413 43.504691 51.173487 664.266095
#> [5111] 16593.904326 282.950405 31.236959 43.570580 20.728093
#> [5116] 43.185005 59.566814 676.587390 16188.642222 313.413631
#> [5121] 38.075133 51.941585 14.116758 49.725098 60.537468
#> [5126] 490.664727 12504.341366 332.336475 37.057482 50.585754
#> [5131] 25.794391 34.584245 39.173538 294.094119 9368.650824
#> [5136] 238.538071 39.816894 53.761216 27.356094 42.350012
#> [5141] 32.642037 161.953584 4959.686799 273.161323 41.500725
#> [5146] 72.503669 22.130488 33.884160 28.541240 80.038212
#> [5151] 1901.186338 188.447495 53.187130 69.078084 33.401141
#> [5156] 33.780515 33.022667 61.955306 1182.946692 215.098218
#> [5161] 42.515117 62.778956 26.216766 35.221615 48.032176
#> [5166] 54.959703 825.698900 207.598380 55.643398 71.384935
#> [5171] 31.635770 35.349741 48.964816 50.205919 578.695492
#> [5176] 265.714340 42.320107 61.631955 24.768630 33.323584
#> [5181] 48.746423 39.307374 432.141679 328.384663 49.987269
#> [5186] 60.807526 28.164686 37.712182 35.494426 33.273868
#> [5191] 327.161537 237.954949 51.004126 62.141679 24.341466
#> [5196] 33.163396 32.038287 57.151888 307.955087 293.975123
#> [5201] 45.686545 60.149132 22.322665 32.619832 39.601874
#> [5206] 23.049419 240.837605 186.115206 46.597708 51.809865
#> [5211] 30.635418 21.868757 36.862236 31.747517 177.808305
#> [5216] 258.993183 35.778968 80.982385 17.748673 31.080975
#> [5221] 27.910608 29.008353 152.458723 206.890375 60.902021
#> [5226] 55.154165 30.905965 35.537998 26.481802 36.547885
#> [5231] 71.281653 319.363948 39.509593 54.674671 23.694966
#> [5236] 22.768565 35.902006 15.519170 122.210194 311.873757
#> [5241] 54.575711 52.414871 43.260145 27.726636 26.703534
#> [5246] 29.005692 49.958104 318.460082 32.422931 65.452027
#> [5251] 16.243843 29.931222 67.388561 21.198202 115.150943
#> [5256] 311.011074 49.097036 34.894409 32.663204 26.605071
#> [5261] 143.219070 132.124666 52.465763 726.977373 24.659910
#> [5266] 50.157988 9.839966 34.117595 213.281965 367.988658
#> [5271] 55.975199 1133.979505 35.030130 48.212218 18.222072
#> [5276] 27.248220 749.983686 568.723259 73.905793 1688.907677
#> [5281] 28.385296 40.115522 9.298693 25.190910 284.538703
#> [5286] 1371.456125 51.937697 1970.014395 31.139139 30.812823
#> [5291] 17.957888 20.077910 2250.042225 247.380044 120.841294
#> [5296] 358.835250 29.951044 30.655786 10.936186 22.041673
#> [5301] 842.268770 684.991122 50.578106 751.022704 17.030382
#> [5306] 56.704069 9.993893 26.125907 395.690440 108.014778
#> [5311] 52.044293 173.997155 27.761691 56.691786 8.355937
#> [5316] 25.420549 121.274068 109.111402 40.842086 185.723421
#> [5321] 19.028397 54.993547 8.221156 30.516583 74.598682
#> [5326] 54.938835 47.603433 208.145478 26.427559 48.410746
#> [5331] 11.010037 29.746014 95.137569 31.070524 62.041933
#> [5336] 81.034690 27.960036 50.010338 13.210586 25.622529
#> [5341] 62.798812 34.743141 57.948189 67.948480 29.345248
#> [5346] 43.847262 10.212893 24.417106 37.715088 24.016516
#> [5351] 51.844956 56.948543 23.039180 47.985173 15.485633
#> [5356] 17.196868 28.604007 28.672823 49.094864 85.515405
#> [5361] 20.712237 54.197844 9.996985 24.535078 30.869278
#> [5366] 33.406760 37.092492 108.928467 29.427018 40.057003
#> [5371] 11.233572 25.663400 26.074822 14.684448 44.153481
#> [5376] 79.863051 20.674375 53.945479 8.635538 21.866742
#> [5381] 21.568681 27.295084 37.782676 119.027245 19.828372
#> [5386] 64.745642 6.866438 37.820110 16.976812 31.202158
#> [5391] 28.064303 126.817124 18.990707 62.140496 7.116460
#> [5396] 29.441396 13.949700 37.203920 24.919200 207.062197
#> [5401] 29.096854 47.315740 14.389914 27.333264 21.161312
#> [5406] 22.028162 24.699609 255.689930 37.234862 35.792454
#> [5411] 19.542832 31.829631 31.247314 19.193122 39.076528
#> [5416] 151.820509 35.495920 47.397914 20.446395 31.425314
#> [5421] 34.012810 15.253335 31.759759 121.802695 31.030318
#> [5426] 45.767372 12.877493 24.125255 27.306708 18.044403
#> [5431] 30.132601 109.521549 41.443886 49.541559 14.060000
#> [5436] 19.674713 30.753903 13.244524 32.146125 147.121849
#> [5441] 37.313982 45.717337 20.340095 23.995267 22.475538
#> [5446] 15.280520 30.612373 126.313107 34.105550 45.470899
#> [5451] 13.900067 23.654804 24.500477 16.061011 30.066356
#> [5456] 144.995349 35.720150 52.332116 22.641242 21.791053
#> [5461] 23.008396 17.488522 26.974505 117.847114 30.733337
#> [5466] 40.035589 13.306863 24.808838 22.640671 11.095066
#> [5471] 37.035494 169.536042 34.706612 42.308956 11.265198
#> [5476] 25.540988 17.534398 13.917723 36.879266 95.253177
#> [5481] 28.148921 54.359477 12.156747 23.301489 18.149539
#> [5486] 9.520780 25.295166 108.913805 31.419713 34.356447
#> [5491] 13.770139 23.333087 18.118756 9.574440 29.141270
#> [5496] 95.353999 35.310113 34.061251 13.283332 22.279662
#> [5501] 17.767520 10.718367 28.965177 93.648825 31.672368
#> [5506] 50.220073 13.240031 19.169652 16.961605 10.676379
#> [5511] 27.901531 101.624876 30.922475 30.711283 10.036392
#> [5516] 23.545922 15.679841 23.386392 32.812637 88.778950
#> [5521] 32.032750 38.288512 11.881548 17.974256 16.239587
#> [5526] 11.168106 24.564803 100.224677 27.833303 57.880141
#> [5531] 9.091872 15.109380 14.008010 10.726497 35.164280
#> [5536] 97.430139 27.099293 37.656634 11.423134 19.898495
#> [5541] 19.736534 10.571789 29.370398 124.398256 27.596925
#> [5546] 33.851647 11.234983 16.486515 15.795668 13.592158
#> [5551] 27.078752 93.896182 31.460622 44.212066 9.682059
#> [5556] 19.535536 14.092844 9.353039 32.686904 79.027967
#> [5561] 28.658147 38.346754 12.294601 14.046168 16.102160
#> [5566] 10.279072 34.637368 52.164251 28.343455 34.719642
#> [5571] 12.085510 16.228113 15.652985 9.625466 32.608459
#> [5576] 50.489848 31.156872 33.528733 10.087299 16.134050
#> [5581] 13.856099 7.972964 36.169426 88.756489 28.420947
#> [5586] 34.800597 9.344251 14.378970 15.781564 6.902752
#> [5591] 31.046286 59.280232 22.718516 32.093968 7.078729
#> [5596] 15.966721 14.978711 8.551211 28.791502 61.168031
#> [5601] 28.751535 34.515249 10.039587 15.328628 13.471790
#> [5606] 11.967418 22.615952 59.452777 25.812509 34.832149
#> [5611] 14.232937 18.010681 13.874654 12.665227 25.134650
#> [5616] 81.158182 25.747741 36.183789 9.222818 13.347133
#> [5621] 18.191310 5.800864 23.016008 59.047435 23.624210
#> [5626] 36.489993 8.455181 13.820350 20.218870 14.415679
#> [5631] 21.961414 42.435816 22.019617 24.294659 8.502477
#> [5636] 14.249668 21.836311 11.240619 31.662255 53.946034
#> [5641] 43.582236 29.396582 10.156057 13.793700 14.460816
#> [5646] 6.621688 23.156595 41.370287 23.171764 39.400401
#> [5651] 6.507159 12.296025 14.287846 7.503581 23.035727
#> [5656] 44.507807 21.540643 29.158486 8.421196 14.577786
#> [5661] 16.518687 6.551813 17.652274 40.726932 21.477399
#> [5666] 27.832757 5.174307 17.541639 15.466425 6.045972
#> [5671] 23.942745 42.047886 20.552341 21.297594 11.327878
#> [5676] 14.112659 13.465211 7.561116 20.891421 39.545105
#> [5681] 19.466234 25.902088 6.970867 10.721802 14.237479
#> [5686] 7.934469 18.993176 43.471168 18.829538 28.251999
#> [5691] 6.619835 14.278923 11.615486 10.125237 25.820873
#> [5696] 37.290990 22.039825 45.526493 8.860670 13.170684
#> [5701] 11.965083 5.701149 22.970239 47.536575 17.859171
#> [5706] 27.403849 11.756828 13.365287 11.563088 6.603249
#> [5711] 19.394457 38.509610 19.594411 27.302006 7.978791
#> [5716] 12.335761 13.157835 5.887464 20.281194 50.624110
#> [5721] 18.197701 23.702696 12.613101 13.236488 15.888507
#> [5726] 6.469216 20.713158 33.497166 21.683398 20.855007
#> [5731] 6.527923 14.409809 12.152113 5.829081 21.829375
#> [5736] 42.967839 18.284162 25.953627 7.302648 15.312105
#> [5741] 11.163051 7.059352 22.451720 55.118277 14.706257
#> [5746] 24.231435 6.239305 11.933300 11.482309 13.922378
#> [5751] 23.312185 35.999993 18.468304 22.806715 6.053295
#> [5756] 13.681849 10.199348 6.919740 21.173732 38.787369
#> [5761] 18.394192 24.134528 5.934901 11.611764 11.985345
#> [5766] 6.137566 25.370443 36.355738 30.076121 24.597549
#> [5771] 8.476769 11.829287 12.237094 7.833082 17.024092
#> [5776] 31.942338 17.740252 26.062882 7.070393 13.139167
#> [5781] 10.057769 5.095920 20.020777 40.341561 15.354534
#> [5786] 29.237354 8.457130 10.475425 10.036429 10.087661
#> [5791] 21.093659 47.750642 17.410353 29.669635 6.080444
#> [5796] 12.988895 10.065906 7.317907 19.790424 30.071568
#> [5801] 26.804742 22.730861 6.864117 10.676785 11.237936
#> [5806] 5.643706 21.637751 43.567709 27.582795 24.688186
#> [5811] 6.651049 12.489531 12.305251 5.027223 21.187252
#> [5816] 39.480930 17.954805 21.496859 7.088664 10.972968
#> [5821] 10.433393 7.064342 17.486392 41.079621 12.462918
#> [5826] 22.563665 4.645000 10.210050 9.841871 4.545358
#> [5831] 18.956592 37.958999 17.593745 23.096646 5.675881
#> [5836] 11.308275 11.962768 6.282951 20.170424 47.077768
#> [5841] 15.466341 23.863601 7.573546 11.824666 10.677047
#> [5846] 4.094874 19.369373 48.449219 16.441603 28.571513
#> [5851] 5.143756 13.317933 10.858247 6.331893 21.963200
#> [5856] 36.229585 15.474986 20.541464 7.247845 12.542505
#> [5861] 11.423010 6.824097 15.968173 39.267411 14.246214
#> [5866] 23.859755 4.786252 10.830720 9.510862 4.685188
#> [5871] 17.486187 32.752970 14.303565 21.148966 7.471369
#> [5876] 11.557634 9.989837 5.051698 19.569410 42.029570
#> [5881] 15.710827 20.697971 6.101037 10.979036 12.173694
#> [5886] 5.356524 18.635946 34.636425 16.274177 22.425319
#> [5891] 7.265970 11.199063 11.075436 5.460401 16.274875
#> [5896] 43.487680 16.894735 22.490648 5.658307 11.431265
#> [5901] 12.308903 3.716979 14.761261 30.137723 26.085530
#> [5906] 23.743736 5.511207 9.487293 11.727344 3.946854
#> [5911] 18.358340 30.042680 17.640732 19.396595 5.540704
#> [5916] 12.829256 9.375596 4.478811 20.907189 31.625138
#> [5921] 14.311768 21.888127 6.201748 10.436704 11.062678
#> [5926] 4.800994 22.197087 37.329104 16.905886 21.342389
#> [5931] 4.184414 9.794905 12.300346 6.322615 19.054385
#> [5936] 25.624018 15.559821 21.976108 5.925293 10.548200
#> [5941] 11.061320 3.928094 21.721847 31.082930 15.730390
#> [5946] 22.552142 4.821667 11.816226 11.516344 5.600983
#> [5951] 19.042766 27.369907 13.867009 19.324885 4.316336
#> [5956] 11.744548 10.201385 6.862863 14.868542 25.671994
#> [5961] 21.497115 19.950111 5.227230 12.352900 11.793891
#> [5966] 7.922085 16.467514 29.233923 19.342515 24.204033
#> [5971] 6.817222 11.201137 13.313019 4.572647 24.195375
#> [5976] 31.381669 17.205520 22.203010 11.207973 10.988817
#> [5981] 8.733691 4.249936 22.672829 27.506185 18.057019
#> [5986] 22.534012 5.698990 10.988898 12.000976 5.925525
#> [5991] 17.584087 36.361166 17.059897 19.463759 5.744939
#> [5996] 14.029562 12.062251 6.391228 20.279620 20.916822
#> [6001] 15.465160 21.549043 6.722870 12.028100 9.949838
#> [6006] 6.007326 24.382272 33.732603 18.271602 21.577365
#> [6011] 6.615802 10.172838 10.803664 3.854277 18.433202
#> [6016] 30.541047 18.613627 21.680726 7.294082 11.106864
#> [6021] 12.282757 5.239440 19.904770 28.836984 15.674173
#> [6026] 21.713432 13.651867 11.300693 10.528669 7.567143
#> [6031] 18.464297 29.124795 14.150920 25.964121 5.663315
#> [6036] 11.118359 11.280843 5.713714 17.904503 36.231586
#> [6041] 21.510322 18.696684 5.041147 9.124287 11.431144
#> [6046] 3.798698 18.065674 25.423382 20.677185 20.894602
#> [6051] 5.702843 10.777248 10.712305 5.173653 20.071802
#> [6056] 36.382167 17.871196 22.533954 6.061124 11.017222
#> [6061] 9.661962 5.581918 18.825902 30.713348 22.574746
#> [6066] 21.728186 6.972377 11.105360 13.780225 4.550884
#> [6071] 19.395026 26.880207 14.210696 22.155957 6.797550
#> [6076] 10.275557 12.222819 8.398578 24.344253 34.193368
#> [6081] 15.504388 20.300883 5.677231 10.249366 12.681867
#> [6086] 4.815232 26.414719 32.685804 19.477807 25.813704
#> [6091] 5.742608 10.393623 11.986271 6.906906 16.276390
#> [6096] 26.596281 13.400807 18.981896 4.858285 10.454178
#> [6101] 12.701189 7.966966 20.260167 30.027440 18.859080
#> [6106] 22.618743 5.682270 12.054238 11.638004 5.021385
#> [6111] 15.539340 34.765216 19.191591 15.998954 5.048621
#> [6116] 9.386482 13.078936 5.873381 15.623255 28.514973
#> [6121] 15.029830 19.101041 6.477837 11.175430 12.146461
#> [6126] 5.148064 18.652995 29.943941 18.033890 23.966590
#> [6131] 6.252118 9.911543 11.501488 3.347896 14.578482
#> [6136] 25.920330 16.644279 17.580480 6.416454 10.302214
#> [6141] 9.645012 6.561954 17.809095 26.193635 16.791367
#> [6146] 25.535732 4.844136 11.837402 8.871229 4.757383
#> [6151] 15.651360 26.396547 14.027239 19.791865 9.792290
#> [6156] 8.804900 10.444221 5.243068 19.937082 26.089301
#> [6161] 18.055032 22.551599 6.192553 9.915538 9.173633
#> [6166] 5.868727 15.541182 24.232333 14.435194 23.942167
#> [6171] 5.017874 10.179691 11.269468 4.221513 20.727558
#> [6176] 26.321338 19.164747 18.945027 7.669707 11.056863
#> [6181] 11.127577 12.666749 15.256717 25.997107 17.438546
#> [6186] 19.986527 6.671356 10.993758 9.369533 4.444676
#> [6191] 21.731148 28.489718 13.770881 18.928200 5.659063
#> [6196] 10.686092 9.026780 4.218146 15.360442 32.986778
#> [6201] 14.450117 23.222753 6.086246 10.253449 10.288777
#> [6206] 5.155154 16.632369 24.452634 15.953815 16.944394
#> [6211] 5.786364 10.837662 12.010598 4.611328 19.639332
#> [6216] 23.316960 16.959275 25.506402 4.360476 10.334268
#> [6221] 11.445835 8.239900 14.574909 25.082319 11.856776
#> [6226] 26.322803 4.254589 11.393935 9.486259 4.610584
#> [6231] 15.952926 27.274178 16.735782 21.962689 5.648849
#> [6236] 12.947390 11.642464 4.671631 15.635235 25.916409
#> [6241] 16.055308 18.728775 6.844440 9.780797 10.753023
#> [6246] 6.802180 21.511615 52.861304 13.085859 21.294226
#> [6251] 5.747175 8.597011 10.058490 8.907762 15.482151
#> [6256] 35.679026 16.521257 27.283316 4.951118 10.781809
#> [6261] 9.012623 6.569064 19.638396 29.239237 13.164390
#> [6266] 23.731186 5.510069 10.607306 13.007705 4.549610
#> [6271] 18.385448 33.727136 14.876893 28.768128 6.202144
#> [6276] 11.445276 11.072589 5.572842 20.476118 25.933766
#> [6281] 15.658919 20.068896 5.536041 10.364386 12.191351
#> [6286] 4.242294 18.140369 28.400607 17.945117 23.952858
#> [6291] 5.254488 9.975148 13.029144 4.996982 16.987497
#> [6296] 21.594442 19.360542 18.129262 6.440756 9.416642
#> [6301] 8.879312 3.974525 22.951163 29.104788 17.868570
#> [6306] 20.328365 11.548576 10.085428 10.498596 4.715607
#> [6311] 24.529314 26.396005 21.096141 19.854503 9.045296
#> [6316] 10.613922 12.959016 5.471734 21.122166 30.976210
#> [6321] 17.467314 30.765345 6.214583 10.425230 9.409511
#> [6326] 5.592456 16.731984 23.036097 17.275026 21.311622
#> [6331] 5.486886 10.735616 9.364954 3.541165 20.435235
#> [6336] 23.682017 12.751667 19.746048 7.011281 9.291310
#> [6341] 8.632754 4.888646 22.854153 42.580133 22.856214
#> [6346] 20.414992 6.439880 11.011290 12.585844 4.953826
#> [6351] 24.834518 30.640564 13.469658 30.635091 6.262156
#> [6356] 11.240475 11.193622 4.392331 19.049030 28.331619
#> [6361] 19.054333 33.136650 5.985691 10.087040 9.629414
#> [6366] 4.252129 17.535644 22.333125 15.536449 20.266349
#> [6371] 5.705797 11.686510 12.066039 6.810281 21.422717
#> [6376] 30.576053 15.644490 18.046062 6.146557 11.416335
#> [6381] 11.477552 5.818372 18.691383 21.365292 13.661480
#> [6386] 19.384176 5.654201 11.051018 10.236746 5.007565
#> [6391] 18.888312 25.496562 12.647561 24.443660 7.740616
#> [6396] 11.405505 11.184417 12.563951 20.612560 27.299467
#> [6401] 16.240234 21.575600 6.522759 10.468161 11.962806
#> [6406] 4.640039 12.203463 20.964559 15.498840 19.165081
#> [6411] 4.929392 10.422037 11.032308 7.748884 19.017662
#> [6416] 28.355905 13.492469 20.585249 6.358037 10.892174
#> [6421] 10.782553 6.424071 20.991071 26.787088 36.758570
#> [6426] 21.156857 5.091073 10.649386 10.809779 5.820364
#> [6431] 18.408146 35.748520 25.736241 19.774131 7.259056
#> [6436] 10.382411 10.373931 4.647639 14.995460 22.978675
#> [6441] 19.219628 22.706139 7.141451 11.857613 10.200689
#> [6446] 5.671289 20.476081 25.707751 17.518600 30.063380
#> [6451] 7.862410 11.423195 12.148871 6.945512 16.150527
#> [6456] 24.298117 14.559476 20.360127 4.588653 11.398603
#> [6461] 9.113533 4.713270 16.622362 31.611712 14.491402
#> [6466] 21.475547 6.315935 11.508246 11.642655 5.202209
#> [6471] 20.508794 30.428672 19.069939 21.935285 6.848964
#> [6476] 11.256043 12.959254 11.712290 17.863851 23.550501
#> [6481] 12.352790 26.415876 5.366100 11.636339 11.313623
#> [6486] 4.908458 18.968952 26.960806 18.474412 23.130283
#> [6491] 6.378123 11.770825 9.588354 9.072638 18.655192
#> [6496] 26.455795 21.032671 31.279063 7.377360 12.570899
#> [6501] 9.846947 4.500947 18.131702 24.298941 17.305334
#> [6506] 20.882561 6.153599 9.618351 10.714424 11.896306
#> [6511] 20.155444 29.094227 15.572138 28.487446 10.424288
#> [6516] 11.914281 12.415575 5.900483 19.919316 22.812075
#> [6521] 15.593424 26.030601 7.840797 11.509692 12.017372
#> [6526] 8.057531 22.592977 22.892289 14.215761 20.443693
#> [6531] 5.393908 11.870577 11.834267 6.053441 15.883934
#> [6536] 23.083281 16.238323 22.422582 6.289417 10.830067
#> [6541] 10.223057 7.172563 17.810912 24.632146 13.679702
#> [6546] 20.500806 6.857304 10.062145 9.713911 6.287977
#> [6551] 14.404737 27.440468 17.232877 26.050061 16.110817
#> [6556] 9.997268 11.447893 4.527345 17.506791 27.246427
#> [6561] 15.694653 20.233884 6.666143 8.928364 10.965052
#> [6566] 3.648313 23.213616 33.352166 15.071790 36.678364
#> [6571] 7.022363 11.270742 8.838623 13.090240 19.128171
#> [6576] 35.436642 19.507580 18.148779 4.615980 9.578203
#> [6581] 10.253686 4.703976 20.169298 22.556414 16.640852
#> [6586] 24.354504 5.193703 12.812498 10.455099 4.696572
#> [6591] 22.974636 27.090029 19.984680 25.796121 7.334822
#> [6596] 11.942260 10.560468 5.169543 20.273639 26.778747
#> [6601] 17.712928 47.051205 5.623750 10.359454 10.664079
#> [6606] 4.895109 19.253780 27.860563 17.694158 28.949455
#> [6611] 8.289831 11.102530 11.607740 3.466563 14.714239
#> [6616] 29.265666 14.398066 22.644254 5.936996 10.778961
#> [6621] 11.092141 4.643042 21.476321 27.218363 17.894117
#> [6626] 23.355493 5.036956 9.768475 10.394807 4.895581
#> [6631] 20.026397 29.728378 14.898927 20.384213 5.368960
#> [6636] 10.511783 10.439251 6.390248 22.113762 28.523886
#> [6641] 13.802355 21.631001 9.026024 11.403928 9.918543
#> [6646] 4.623812 15.742373 28.029476 18.554355 22.773872
#> [6651] 5.746443 11.767112 10.083992 4.374796 17.626457
#> [6656] 27.991132 15.331903 23.400512 6.216858 12.313058
#> [6661] 10.803967 5.525187 15.587872 23.094022 15.856851
#> [6666] 18.961042 6.198267 11.592470 10.174680 4.796777
#> [6671] 19.315707 27.416641 15.184901 24.891714 6.427024
#> [6676] 10.255672 11.831430 5.093051 20.173487 27.496317
#> [6681] 20.103094 38.396415 6.865835 11.102031 11.310023
#> [6686] 5.478815 18.663656 24.478150 16.901445 21.618265
#> [6691] 7.999599 10.073415 7.744202 8.749388 19.594045
#> [6696] 26.903182 14.525386 22.084893 5.142531 11.084978
#> [6701] 10.620466 7.738099 18.321208 29.081020 13.301647
#> [6706] 19.992725 5.895386 10.644138 11.859441 7.282677
#> [6711] 18.018021 28.188249 24.760736 22.038372 4.366667
#> [6716] 11.001688 12.499550 5.123093 15.262302 23.756095
#> [6721] 20.720150 30.228790 4.394520 10.028479 9.707609
#> [6726] 4.350069 18.140195 20.009206 17.993648 19.002447
#> [6731] 6.299981 11.473525 9.543563 4.155215 15.328477
#> [6736] 28.991882 17.768750 22.615224 6.139764 11.501982
#> [6741] 13.734892 5.499403 16.914373 28.790825 16.872875
#> [6746] 18.492110 5.583537 8.935665 11.769684 5.286785
#> [6751] 18.034496 32.084175 15.515344 19.329474 8.072934
#> [6756] 11.121037 9.593857 5.412797 19.771129 23.106614
#> [6761] 17.579322 20.187170 5.363943 11.451182 11.587418
#> [6766] 5.364188 16.048256 25.325948 15.721866 18.614385
#> [6771] 6.805010 9.811136 11.751012 3.874564 23.567623
#> [6776] 23.543039 13.171079 27.936709 8.197528 11.255197
#> [6781] 8.107489 7.452791 21.564299 27.342121 12.945750
#> [6786] 25.453192 6.986556 9.905569 9.395154 4.332932
#> [6791] 16.783253 22.894339 24.797071 22.335369 6.891753
#> [6796] 11.678509 10.689191 4.912379 21.466754 23.509351
#> [6801] 13.218125 17.953736 8.571553 9.593539 10.262257
#> [6806] 6.482088 16.753287 34.789242 15.390476 22.116460
#> [6811] 6.477632 10.788972 13.416375 5.291164 21.663222
#> [6816] 32.065756 14.023479 30.832228 5.314142 9.250391
#> [6821] 9.214860 6.348743 18.339258 24.655968 13.249055
#> [6826] 55.532747 6.780604 11.377478 13.323924 6.216104
#> [6831] 14.935224 25.768693 12.909203 22.671930 4.488649
#> [6836] 10.799970 10.937674 5.255813 19.616507 23.172698
#> [6841] 15.823211 21.245194 7.096178 9.550661 11.152417
#> [6846] 8.152236 21.084419 26.770305 14.429645 21.271298
#> [6851] 7.832856 12.056087 11.779300 4.475797 18.643286
#> [6856] 26.387191 14.730316 36.594233 8.089238 11.029222
#> [6861] 10.422804 7.267043 15.095851 26.593623 21.754364
#> [6866] 23.551933 5.089353 10.360865 11.938029 5.753962
#> [6871] 20.659532 24.610320 21.376482 20.406358 5.275592
#> [6876] 9.250746 10.613810 3.960601 17.330385 30.206389
#> [6881] 15.360810 29.016053 5.530230 11.171647 9.412464
#> [6886] 5.113704 21.928675 38.104277 11.901806 21.775210
#> [6891] 5.200633 10.539019 10.844053 6.163663 16.175943
#> [6896] 36.678962 16.282467 38.056857 5.362595 11.323132
#> [6901] 10.703461 5.397676 15.702316 24.359320 15.139917
#> [6906] 18.602421 4.762504 10.856186 14.184799 8.808151
#> [6911] 14.704503 31.305581 11.804618 26.150565 6.763011
#> [6916] 9.497258 11.582165 5.394998 23.759381 20.320550
#> [6921] 15.241552 16.246039 4.684850 10.979298 10.456820
#> [6926] 8.905588 14.781592 27.012876 14.292981 33.032637
#> [6931] 5.100140 10.400169 13.934594 4.795497 17.293917
#> [6936] 20.621456 13.904029 17.864621 5.156797 10.480541
#> [6941] 10.470159 4.898430 14.563778 25.385020 21.642064
#> [6946] 18.811826 4.960947 11.097639 13.182356 5.667703
#> [6951] 18.891917 21.366534 12.954209 22.733060 7.033172
#> [6956] 11.417416 9.605428 5.253914 19.270777 25.843744
#> [6961] 14.485716 25.597451 5.316602 10.640504 9.586580
#> [6966] 5.366495 13.176101 30.195574 18.606871 17.343704
#> [6971] 7.126794 9.636645 12.942500 4.748871 18.303082
#> [6976] 23.545868 16.706177 25.737615 6.227998 11.098120
#> [6981] 10.200699 6.050280 18.225609 27.069885 14.260101
#> [6986] 19.685704 5.140146 12.979811 9.685239 3.892039
#> [6991] 19.996609 28.714788 12.774022 20.914691 5.563148
#> [6996] 11.257266 7.818056 8.191035 19.670319 24.455115
#> [7001] 13.901029 20.666881 5.740455 10.390315 10.540737
#> [7006] 5.239556 18.002698 30.751921 14.245768 22.747782
#> [7011] 6.905547 11.496915 12.349256 6.612709 18.463281
#> [7016] 28.900459 15.671822 19.157494 7.323224 10.722297
#> [7021] 11.034751 10.277040 21.406327 29.377878 14.209451
#> [7026] 20.876081 5.839155 11.080671 10.919747 5.575396
#> [7031] 15.760910 26.419115 13.416710 16.347512 5.374161
#> [7036] 9.481983 12.547474 4.060674 17.018011 30.737735
#> [7041] 13.810171 17.649129 5.622687 10.487182 10.083001
#> [7046] 6.024245 15.634883 30.548391 25.156382 29.441643
#> [7051] 8.865323 12.989413 12.099445 4.575110 15.880972
#> [7056] 28.032077 15.624855 26.689582 7.208590 9.815443
#> [7061] 10.065303 3.596404 18.388266 28.468746 15.937781
#> [7066] 18.162128 6.642714 10.818792 13.257751 3.985955
#> [7071] 15.419146 26.977868 13.988382 17.933015 8.956976
#> [7076] 10.258652 12.037752 3.863613 18.960030 32.534989
#> [7081] 14.185539 16.000703 6.372031 11.121082 12.152146
#> [7086] 5.688608 17.753208 30.389682 15.119966 19.452684
#> [7091] 8.024150 10.194372 10.160811 7.108232 17.640853
#> [7096] 25.193212 13.075439 16.072879 6.290957 9.592162
#> [7101] 12.689229 4.033864 20.406621 23.419240 18.145626
#> [7106] 21.302784 5.481212 11.696416 11.580833 4.831010
#> [7111] 22.675253 32.060353 16.695246 25.414814 6.109536
#> [7116] 11.846644 10.857186 5.205461 16.780919 25.427510
#> [7121] 16.107229 17.122453 5.059080 10.556342 11.822079
#> [7126] 4.515281 14.988842 30.244731 12.690808 19.665574
#> [7131] 6.713899 9.560162 9.421713 5.681562 16.055999
#> [7136] 28.171817 20.712684 20.664815 6.748661 12.571693
#> [7141] 8.823148 5.822341 20.121793 20.979256 18.631864
#> [7146] 21.673262 7.629184 13.502819 9.073333 8.684805
#> [7151] 16.186168 21.464304 15.091964 19.862444 4.927465
#> [7156] 12.930948 14.048988 7.158971 17.242263 23.563460
#> [7161] 15.466045 19.277646 7.587924 10.872017 9.533815
#> [7166] 4.970147 16.913882 24.449818 43.758709 25.863644
#> [7171] 5.232467 9.738889 11.804419 8.489460 16.669560
#> [7176] 26.688257 17.219719 27.996507 5.178023 11.167806
#> [7181] 11.004149 4.800555 19.655563 43.036808 15.986097
#> [7186] 18.054808 5.628900 11.407267 10.183817 4.532392
#> [7191] 20.219760 27.134075 13.048674 21.588818 5.518193
#> [7196] 12.782701 9.393076 5.146381 18.904115 22.286011
#> [7201] 18.671808 24.223806 8.796984 11.846353 13.320911
#> [7206] 6.489286 19.517095 29.724709 10.712072 17.545316
#> [7211] 4.368678 8.627120 10.559412 5.426895 17.799580
#> [7216] 25.874331 17.997040 18.517430 5.618141 11.743682
#> [7221] 11.158027 3.575223 20.657490 32.562441 19.037497
#> [7226] 26.273791 6.283998 12.640235 11.624089 4.913197
#> [7231] 20.571886 30.399636 15.870522 24.490590 6.410994
#> [7236] 13.209832 11.364761 5.087315 16.052558 22.931022
#> [7241] 16.044677 31.339568 6.483887 11.570144 10.642505
#> [7246] 11.810883 20.532468 24.897924 14.516260 19.030195
#> [7251] 5.798249 10.029972 9.873241 3.567322 15.658822
#> [7256] 23.023953 13.123244 23.926514 4.117592 10.599039
#> [7261] 10.300942 3.321613 20.175308 23.785457 14.509117
#> [7266] 19.682312 4.608440 10.591288 9.225198 8.423284
#> [7271] 22.571393 22.472397 14.747287 19.000214 5.375725
#> [7276] 11.626566 13.137880 4.952970 19.718954 26.383357
#> [7281] 15.215226 28.117391 5.542641 12.041772 11.480610
#> [7286] 5.272277 20.217408 34.523173 15.138853 27.339873
#> [7291] 4.612850 11.663930 10.295282 5.648234 16.710742
#> [7296] 26.140353 16.727275 22.147068 5.179146 10.149677
#> [7301] 10.860208 4.961367 18.097953 27.945734 18.120679
#> [7306] 21.437620 6.141739 10.819232 9.853445 5.869381
#> [7311] 15.428073 27.692158 19.264237 27.566039 6.393489
#> [7316] 10.265059 10.295941 3.146296 14.911231 27.209805
#> [7321] 17.108452 22.307817 5.526573 11.341389 10.502163
#> [7326] 5.046900 23.287188 26.458952 18.182431 15.872881
#> [7331] 4.926028 10.896387 9.224376 6.180670 17.732998
#> [7336] 21.434360 18.239937 20.242460 6.465121 12.215855
#> [7341] 10.073714 7.710437 19.436794 34.905662 18.347346
#> [7346] 24.781970 4.619737 10.903837 9.441030 2.893308
#> [7351] 16.659490 39.575581 14.102785 22.814500 5.316469
#> [7356] 13.326233 10.705395 5.415616 20.063398 23.376480
#> [7361] 17.464293 21.574140 4.836159 11.063343 9.073998
#> [7366] 5.139404 20.440055 32.329492 18.073725 24.193438
#> [7371] 5.621191 11.747614 10.044469 4.964368 21.480612
#> [7376] 24.070078 18.499838 25.460212 5.770030 11.065400
#> [7381] 11.097140 5.494908 20.947693 20.181266 14.691009
#> [7386] 18.358518 12.839608 10.779411 11.778241 4.042762
#> [7391] 14.593209 24.424841 18.810647 30.670111 5.781123
#> [7396] 12.204886 12.370435 7.702345 19.604917 27.572259
#> [7401] 14.847023 22.105461 5.941954 11.608932 11.650530
#> [7406] 4.810545 15.205574 27.321689 15.771732 29.597042
#> [7411] 5.330957 10.728306 11.132029 11.893980 19.501164
#> [7416] 25.440793 11.841345 18.703986 5.092588 13.080661
#> [7421] 11.049942 13.596787 16.725051 28.551876 18.751091
#> [7426] 20.968913 5.036960 10.868268 11.691249 5.074553
#> [7431] 19.074393 20.464564 13.393238 22.127389 5.214133
#> [7436] 11.972148 10.633052 5.715829 17.524953 30.918745
#> [7441] 17.044198 20.282594 5.437108 12.423008 11.990285
#> [7446] 4.790931 19.447584 29.974709 19.251738 17.455773
#> [7451] 6.751485 11.323130 10.531420 7.612166 21.257715
#> [7456] 20.800256 13.720112 25.410774 4.919325 10.800045
#> [7461] 9.372462 5.072446 20.185149 38.150772 19.376895
#> [7466] 20.854240 5.337673 11.381572 12.616399 3.990814
#> [7471] 18.291332 25.068256 12.631867 23.330574 6.869057
#> [7476] 12.553170 11.815984 5.021365 20.802859 28.155576
#> [7481] 15.764191 26.821764 6.360442 11.766035 10.782271
#> [7486] 3.896033 17.382828 27.710311 18.100761 17.079317
#> [7491] 7.684174 11.574225 10.768669 8.289828 18.917753
#> [7496] 26.804411 13.688045 21.537911 5.078491 12.180277
#> [7501] 10.125505 5.288098 22.262421 24.846690 21.931702
#> [7506] 23.686373 6.074599 11.436642 10.600646 4.518934
#> [7511] 21.997900 21.524193 23.527411 26.759419 7.094081
#> [7516] 12.090066 10.643323 4.132045 18.537347 19.476560
#> [7521] 25.686040 21.352789 6.128181 12.199460 13.321196
#> [7526] 2.734434 21.206933 23.053278 17.372551 25.798210
#> [7531] 9.698968 14.378929 12.645599 7.810056 20.034915
#> [7536] 32.555793 16.791938 25.004879 5.251820 13.702863
#> [7541] 10.538479 5.009352 22.555193 19.712689 26.936604
#> [7546] 17.422569 6.176965 14.143801 12.003666 3.828041
#> [7551] 17.644515 25.042951 16.887221 30.769232 9.324177
#> [7556] 12.406835 12.606379 5.283516 18.386701 29.474921
#> [7561] 15.251721 19.601222 10.085241 9.814753 10.810465
#> [7566] 4.140590 19.163609 20.468277 14.263511 22.320248
#> [7571] 5.150916 10.569625 11.878047 6.988212 17.878611
#> [7576] 26.540782 15.207089 20.221743 14.457465 11.367417
#> [7581] 11.208363 4.907654 21.556411 30.568173 14.046807
#> [7586] 19.669251 6.402272 13.619962 10.007125 5.780530
#> [7591] 23.186570 27.099030 15.929814 31.258148 7.509852
#> [7596] 9.551942 13.551723 5.922459 16.579424 20.662296
#> [7601] 19.112092 25.091589 4.978148 11.568894 12.625133
#> [7606] 4.354787 23.936968 26.683195 14.458707 21.009509
#> [7611] 12.612336 9.856103 11.225819 8.167260 16.837622
#> [7616] 20.325522 16.058894 20.026479 5.190318 12.508379
#> [7621] 11.215948 4.217643 19.692705 29.805327 15.671152
#> [7626] 18.085552 5.073741 12.106583 11.490157 5.347962
#> [7631] 18.824891 49.726941 14.281069 20.817688 8.415164
#> [7636] 9.796088 10.595431 5.451450 16.636279 31.143978
#> [7641] 19.292997 16.612393 4.891810 11.551406 12.025671
#> [7646] 3.924839 19.266377 22.736391 16.075325 30.839272
#> [7651] 5.590718 13.171760 11.567755 6.094387 16.769438
#> [7656] 28.060955 12.635570 18.827009 4.027460 12.082643
#> [7661] 9.984518 4.672091 24.819565 30.471918 16.111701
#> [7666] 22.382919 4.645265 12.025229 13.000555 5.091949
#> [7671] 26.920995 26.348043 19.936282 24.945016 6.827643
#> [7676] 11.023471 10.900113 8.315678 21.473304 27.530558
#> [7681] 12.454847 21.831697 5.573722 10.601059 11.925730
#> [7686] 5.746961 20.713728 25.798588 16.012434 21.796169
#> [7691] 6.417882 10.700514 10.903377 4.480478 17.149674
#> [7696] 23.878620 18.499575 25.481645 6.656719 13.782775
#> [7701] 12.623189 5.521048 25.624412 31.446268 16.421929
#> [7706] 20.429872 4.733633 10.520906 11.185344 4.420251
#> [7711] 22.497928 36.608600 15.347308 19.480339 7.083301
#> [7716] 10.731928 14.869575 5.547368 18.916802 25.658793
#> [7721] 15.953297 25.185723 7.185226 12.205838 11.457208
#> [7726] 8.220383 17.299302 23.415723 14.061044 20.435312
#> [7731] 7.046283 12.713612 9.526309 7.821084 19.330632
#> [7736] 30.619985 12.920738 25.131478 6.298253 11.351445
#> [7741] 11.885363 4.346974 20.596821 34.553287 15.620047
#> [7746] 18.851762 3.972778 13.309963 7.983911 4.375903
#> [7751] 17.639627 30.102932 111.255685 107.742982 39.323458
#> [7756] 90.245908 41.333908 34.393383 60.231789 1347.062580
#> [7761] 258.993888 149.097302 80.886770 183.057364 380.588923
#> [7766] 221.053078 231.076300 2751.860237 427.376555 220.757567
#> [7771] 181.393390 275.608580 891.967851 372.613964 324.520833
#> [7776] 2654.088363 605.403913 261.982388 205.642529 331.290232
#> [7781] 737.029419 389.073368 315.570563 2277.503047 615.309312
#> [7786] 300.432175 252.564125 404.578241 908.647718 649.023116
#> [7791] 423.792728 1707.197354 827.826970 349.325771 352.824885
#> [7796] 457.667557 702.284586 429.008773 344.530821 2202.876764
#> [7801] 768.317060 465.955844 290.023176 385.290865 623.483698
#> [7806] 402.191514 305.123658 1714.732878 588.782495 461.701825
#> [7811] 247.541897 320.326130 489.561579 343.556420 303.208162
#> [7816] 1539.537585 651.452719 378.687139 248.363507 302.456540
#> [7821] 429.367335 317.360105 250.107659 1538.783435 509.186961
#> [7826] 391.489768 233.971815 260.377352 347.627022 264.328962
#> [7831] 207.426240 1365.394803 523.692695 327.306901 192.844284
#> [7836] 256.843326 362.948637 242.274251 193.427337 1348.491000
#> [7841] 479.303437 285.394727 165.414905 245.095873 336.400719
#> [7846] 217.795558 192.110785 1252.299473 468.573697 439.146607
#> [7851] 186.816167 218.680490 299.378622 222.052751 172.299356
#> [7856] 1210.765626 476.897979 355.980335 183.315618 236.212957
#> [7861] 271.984407 213.512437 180.891237 1179.405691 434.962204
#> [7866] 293.513382 183.294823 251.041310 263.574969 198.637262
#> [7871] 170.918953 1072.756967 440.406159 270.230960 185.138421
#> [7876] 219.393022 234.354640 167.557228 145.656116 1003.720204
#> [7881] 420.371526 280.616540 167.380158 204.790994 212.103685
#> [7886] 159.726511 136.725763 924.848811 454.933102 316.368792
#> [7891] 160.482277 185.209258 230.637496 156.850222 136.952103
#> [7896] 978.977708 408.307152 260.772104 162.477973 180.792609
#> [7901] 185.818480 126.458559 124.354105 924.736240 417.284080
#> [7906] 257.575590 154.447681 168.303761 162.235745 132.724399
#> [7911] 122.158344 858.140658 378.140355 305.451603 135.094730
#> [7916] 161.287850 158.749598 119.737923 117.369899 802.067620
#> [7921] 403.725657 232.105776 126.496602 159.704853 146.799901
#> [7926] 115.072374 103.631570 706.194285 334.775766 261.211185
#> [7931] 128.103187 149.964394 152.966866 117.347215 104.952157
#> [7936] 706.182840 344.046077 269.141127 123.395546 124.171349
#> [7941] 138.099311 82.074642 75.826916 589.043040 223.601279
#> [7946] 175.886064 142.444739 142.247683 111.930238 91.713923
#> [7951] 86.087973 595.800695 369.323350 212.722680 110.991849
#> [7956] 116.459330 129.801881 89.286497 82.584815 543.322802
#> [7961] 315.849776 218.248313 113.947610 121.421001 123.501765
#> [7966] 85.558560 82.558582 641.804290 345.811973 162.071403
#> [7971] 66.968154 141.820448 90.956242 80.273137 68.719788
#> [7976] 414.186888 184.411137 252.840866 110.139158 137.705735
#> [7981] 100.290282 50.685694 89.180058 497.646742 134.601899
#> [7986] 297.016990 85.664223 89.164163 61.245065 95.795605
#> [7991] 82.820765 534.519704 344.328371 196.745896 104.924516
#> [7996] 62.622743 113.532331 85.375663 82.439499 342.765803
## First we list available isolation windows
table(isolationWindowTargetMz(spectra(mse_dia)))
#>
#> 163.75 208.95 244.05 270.85 299.1 329.8 367.35 601.85
#> 1000 1000 1000 1000 1000 1000 1000 1000
## We can then extract the TIC of MS2 data for a specific isolation window
chr_ms2 <- chromatogram(mse_dia, msLevel = 2L,
isolationWindowTargetMz = 244.05)
plot(chr_ms2)
####
## Chromatographic peak detection
## Perform peak detection on the data using the centWave algorith. Note
## that the parameters are chosen to reduce the run time of the example.
p <- CentWaveParam(noise = 10000, snthresh = 40, prefilter = c(3, 10000))
xmse <- findChromPeaks(mse, param = p)
xmse
#> Object of class XcmsExperiment
#> Spectra: MS1 (3834)
#> Experiment data: 3 sample(s)
#> Sample data links:
#> - spectra: 3 sample(s) to 3834 element(s).
#> xcms results:
#> - chromatographic peaks: 248 in MS level(s): 1
## Have a quick look at the identified chromatographic peaks
head(chromPeaks(xmse))
#> mz mzmin mzmax rt rtmin rtmax into intb maxo
#> CP001 453.2 453.2 453.2 2506.073 2501.378 2527.982 1007409.0 1007380.8 38152
#> CP002 302.0 302.0 302.0 2617.185 2595.275 2640.659 687146.6 671297.8 30552
#> CP003 344.0 344.0 344.0 2679.783 2646.919 2709.517 5210015.9 5135916.9 152320
#> CP004 430.1 430.1 430.1 2681.348 2639.094 2712.647 2395840.3 2299899.6 65752
#> CP005 366.0 366.0 366.0 2679.783 2642.224 2718.907 3365174.0 3279468.3 79928
#> CP006 343.0 343.0 343.0 2678.218 2637.529 2712.647 24147443.2 23703761.7 672064
#> sn sample
#> CP001 38151 1
#> CP002 46 1
#> CP003 68 1
#> CP004 42 1
#> CP005 49 1
#> CP006 87 1
## Extract chromatographic peaks identified between 3000 and 3300 seconds
chromPeaks(xmse, rt = c(3000, 3300), type = "within")
#> mz mzmin mzmax rt rtmin rtmax into intb
#> CP021 453.2 453.2 453.2 3063.196 3035.027 3114.840 3001594.78 3001514.97
#> CP030 361.1 361.1 361.1 3147.704 3141.444 3153.964 367361.84 362278.28
#> CP031 340.3 340.3 340.3 3230.646 3225.952 3233.776 68671.25 68664.99
#> CP032 526.2 526.2 526.2 3243.166 3238.471 3246.296 374698.56 374692.30
#> CP033 313.1 313.1 313.1 3276.030 3254.121 3290.115 1199152.63 1162839.05
#> CP034 454.1 454.1 454.1 3276.030 3261.945 3294.809 12283448.95 12024951.77
#> CP108 307.1 307.1 307.1 3143.009 3121.099 3164.918 2191519.65 2063202.03
#> CP109 278.1 278.1 278.1 3196.217 3180.568 3213.432 657840.25 651048.36
#> CP110 526.1 526.1 526.1 3179.003 3150.833 3210.302 21334966.98 21147392.01
#> CP111 380.1 380.1 380.1 3152.398 3132.054 3174.308 2201517.91 2201474.09
#> CP112 380.1 380.1 380.1 3210.302 3210.302 3216.561 187031.44 187023.61
#> CP113 380.1 380.1 380.1 3202.477 3174.308 3216.561 1156780.73 1156736.91
#> CP114 380.1 380.1 380.1 3222.821 3216.561 3240.036 485818.25 485793.21
#> CP115 286.2 286.2 286.2 3258.815 3246.296 3280.725 1264118.64 1247233.70
#> CP116 308.1 308.1 308.1 3261.945 3241.601 3285.419 2066854.37 2025917.82
#> CP204 380.1 380.1 380.1 3150.835 3128.925 3171.179 2036803.99 1937655.24
#> CP205 286.2 286.2 286.2 3250.992 3233.777 3257.252 732016.23 722185.86
#> CP206 568.2 568.2 568.2 3207.173 3185.264 3232.212 3951832.25 3866503.74
#> maxo sn sample
#> CP021 53096 53095 1
#> CP030 49240 57 1
#> CP031 11391 11390 1
#> CP032 60800 60799 1
#> CP033 55392 50 1
#> CP034 554112 58 1
#> CP108 101400 43 2
#> CP109 30208 54 2
#> CP110 622144 251 2
#> CP111 95176 95175 2
#> CP112 25144 28791 2
#> CP113 28920 28919 2
#> CP114 23176 23535 2
#> CP115 67600 41 2
#> CP116 96272 78 2
#> CP204 82624 49 3
#> CP205 74824 68 3
#> CP206 164352 113 3
## Extract ion chromatograms (EIC) for the first two chromatographic
## peaks.
chrs <- chromatogram(xmse,
mz = chromPeaks(xmse)[1:2, c("mzmin", "mzmax")],
rt = chromPeaks(xmse)[1:2, c("rtmin", "rtmax")])
#> Extracting chromatographic data
#> Processing chromatographic peaks
## An EIC for each sample and each of the two regions was extracted.
## Identified chromatographic peaks in the defined regions are extracted
## as well.
chrs
#> XChromatograms with 2 rows and 3 columns
#> ko15.CDF ko16.CDF ko18.CDF
#> <XChromatogram> <XChromatogram> <XChromatogram>
#> [1,] peaks: 1 peaks: 0 peaks: 0
#> [2,] peaks: 1 peaks: 0 peaks: 0
#> phenoData with 3 variables
#> featureData with 4 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
## Plot the EICs for the second defined region
plot(chrs[2, ])
 ## Subsetting the data to the results (and data) for the second sample
a <- xmse[2]
nrow(chromPeaks(xmse))
#> [1] 248
nrow(chromPeaks(a))
#> [1] 100
## Filtering the result by retention time: keeping all spectra and
## chromatographic peaks within 3000 and 3500 seconds.
xmse_sub <- filterRt(xmse, rt = c(3000, 3500))
#> Filter spectra
xmse_sub
#> Object of class XcmsExperiment
#> Spectra: MS1 (960)
#> Experiment data: 3 sample(s)
#> Sample data links:
#> - spectra: 3 sample(s) to 960 element(s).
#> xcms results:
#> - chromatographic peaks: 79 in MS level(s): 1
nrow(chromPeaks(xmse_sub))
#> [1] 79
## Perform an initial feature grouping to allow alignment using the
## peak groups method:
pdp <- PeakDensityParam(sampleGroups = rep(1, 3))
xmse <- groupChromPeaks(xmse, param = pdp)
## Perform alignment using the peak groups method.
pgp <- PeakGroupsParam(span = 0.4)
xmse <- adjustRtime(xmse, param = pgp)
#> Performing retention time alignment using 19 anchor peaks.
## Visualizing the alignment results
plotAdjustedRtime(xmse)
## Performing the final correspondence analysis
xmse <- groupChromPeaks(xmse, param = pdp)
## Show the definition of the first 6 features
featureDefinitions(xmse) |> head()
#> mzmed mzmin mzmax rtmed rtmin rtmax npeaks 1 peakidx
#> FT01 279.0 279.0 279.0 2789.588 2787.430 2791.746 2 2 11, 199
#> FT02 286.2 286.2 286.2 3253.923 3245.811 3262.034 2 2 115, 205
#> FT03 300.2 300.2 300.2 3385.835 3384.068 3390.895 4 3 35, 125,....
#> FT04 301.0 301.0 301.0 2789.066 2787.430 2790.180 3 3 10, 97, 198
#> FT05 305.1 305.1 305.1 2927.922 2922.158 2933.686 2 2 14, 202
#> FT06 305.1 305.1 305.1 3000.543 2991.470 3009.616 2 2 15, 203
#> ms_level
#> FT01 1
#> FT02 1
#> FT03 1
#> FT04 1
#> FT05 1
#> FT06 1
## Extract the feature values; show the results for the first 6 rows.
featureValues(xmse) |> head()
#> ko15.CDF ko16.CDF ko18.CDF
#> FT01 17140627.0 NA 16919266.9
#> FT02 NA 1264119 732016.2
#> FT03 4700903.2 5313736 5169558.2
#> FT04 3051847.8 1964444 2774885.3
#> FT05 1070389.9 NA 1983342.5
#> FT06 847473.1 NA 1003750.6
## The full results can also be extracted as a `SummarizedExperiment`
## that would eventually simplify subsequent analyses with other packages.
## Any additional parameters passed to the function are passed to the
## `featureValues` function that is called to generate the feature value
## matrix.
se <- quantify(xmse, method = "sum")
## EICs for all features can be extracted with the `featureChromatograms`
## function. Note that, depending on the data set, extracting this for
## all features might take some time. Below we extract EICs for the
## first 10 features by providing the feature IDs.
chrs <- featureChromatograms(xmse,
features = rownames(featureDefinitions(xmse))[1:10])
#> Processing chromatographic peaks for features
chrs
#> XChromatograms with 10 rows and 3 columns
#> ko15.CDF ko16.CDF ko18.CDF
#> <XChromatogram> <XChromatogram> <XChromatogram>
#> [1,] peaks: 1 peaks: 0 peaks: 1
#> [2,] peaks: 0 peaks: 1 peaks: 1
#> ... ... ... ...
#> [9,] peaks: 1 peaks: 2 peaks: 0
#> [10,] peaks: 1 peaks: 1 peaks: 1
#> phenoData with 3 variables
#> featureData with 4 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
#> Correspondence:
#> method: chromatographic peak density
#> 10 feature(s) identified.
plot(chrs[3, ])
## Subsetting the data to the results (and data) for the second sample
a <- xmse[2]
nrow(chromPeaks(xmse))
#> [1] 248
nrow(chromPeaks(a))
#> [1] 100
## Filtering the result by retention time: keeping all spectra and
## chromatographic peaks within 3000 and 3500 seconds.
xmse_sub <- filterRt(xmse, rt = c(3000, 3500))
#> Filter spectra
xmse_sub
#> Object of class XcmsExperiment
#> Spectra: MS1 (960)
#> Experiment data: 3 sample(s)
#> Sample data links:
#> - spectra: 3 sample(s) to 960 element(s).
#> xcms results:
#> - chromatographic peaks: 79 in MS level(s): 1
nrow(chromPeaks(xmse_sub))
#> [1] 79
## Perform an initial feature grouping to allow alignment using the
## peak groups method:
pdp <- PeakDensityParam(sampleGroups = rep(1, 3))
xmse <- groupChromPeaks(xmse, param = pdp)
## Perform alignment using the peak groups method.
pgp <- PeakGroupsParam(span = 0.4)
xmse <- adjustRtime(xmse, param = pgp)
#> Performing retention time alignment using 19 anchor peaks.
## Visualizing the alignment results
plotAdjustedRtime(xmse)
## Performing the final correspondence analysis
xmse <- groupChromPeaks(xmse, param = pdp)
## Show the definition of the first 6 features
featureDefinitions(xmse) |> head()
#> mzmed mzmin mzmax rtmed rtmin rtmax npeaks 1 peakidx
#> FT01 279.0 279.0 279.0 2789.588 2787.430 2791.746 2 2 11, 199
#> FT02 286.2 286.2 286.2 3253.923 3245.811 3262.034 2 2 115, 205
#> FT03 300.2 300.2 300.2 3385.835 3384.068 3390.895 4 3 35, 125,....
#> FT04 301.0 301.0 301.0 2789.066 2787.430 2790.180 3 3 10, 97, 198
#> FT05 305.1 305.1 305.1 2927.922 2922.158 2933.686 2 2 14, 202
#> FT06 305.1 305.1 305.1 3000.543 2991.470 3009.616 2 2 15, 203
#> ms_level
#> FT01 1
#> FT02 1
#> FT03 1
#> FT04 1
#> FT05 1
#> FT06 1
## Extract the feature values; show the results for the first 6 rows.
featureValues(xmse) |> head()
#> ko15.CDF ko16.CDF ko18.CDF
#> FT01 17140627.0 NA 16919266.9
#> FT02 NA 1264119 732016.2
#> FT03 4700903.2 5313736 5169558.2
#> FT04 3051847.8 1964444 2774885.3
#> FT05 1070389.9 NA 1983342.5
#> FT06 847473.1 NA 1003750.6
## The full results can also be extracted as a `SummarizedExperiment`
## that would eventually simplify subsequent analyses with other packages.
## Any additional parameters passed to the function are passed to the
## `featureValues` function that is called to generate the feature value
## matrix.
se <- quantify(xmse, method = "sum")
## EICs for all features can be extracted with the `featureChromatograms`
## function. Note that, depending on the data set, extracting this for
## all features might take some time. Below we extract EICs for the
## first 10 features by providing the feature IDs.
chrs <- featureChromatograms(xmse,
features = rownames(featureDefinitions(xmse))[1:10])
#> Processing chromatographic peaks for features
chrs
#> XChromatograms with 10 rows and 3 columns
#> ko15.CDF ko16.CDF ko18.CDF
#> <XChromatogram> <XChromatogram> <XChromatogram>
#> [1,] peaks: 1 peaks: 0 peaks: 1
#> [2,] peaks: 0 peaks: 1 peaks: 1
#> ... ... ... ...
#> [9,] peaks: 1 peaks: 2 peaks: 0
#> [10,] peaks: 1 peaks: 1 peaks: 1
#> phenoData with 3 variables
#> featureData with 4 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
#> Correspondence:
#> method: chromatographic peak density
#> 10 feature(s) identified.
plot(chrs[3, ])