Extract ion chromatograms for each feature
Source:R/AllGenerics.R, R/XcmsExperiment.R, R/methods-XCMSnExp.R
featureChromatograms.RdExtract ion chromatograms for features in an XcmsExperiment or
XCMSnExp object. The function returns for each feature the
extracted ion chromatograms (along with all associated chromatographic
peaks) in each sample. The chromatogram is extracted from the m/z - rt
region that includes all chromatographic peaks of a feature. By default,
this region is defined using the range of the chromatographic peaks' m/z
and retention times (with mzmin = min, mzmax = max, rtmin = min and
rtmax = max). For some features, and depending on the data, the m/z and
rt range can thus be relatively large. The boundaries of the m/z - rt
region can also be restricted by changing parameters mzmin, mzmax,
rtmin and rtmax to a different functions, such as median.
By default only chromatographic peaks associated with a feature are
included in the returned XChromatograms object. For object being an
XCMSnExp object parameter include allows also to return all
chromatographic peaks with their apex position within the selected
region (include = "apex_within") or any chromatographic peak overlapping
the m/z and retention time range (include = "any").
Usage
featureChromatograms(object, ...)
# S4 method for class 'XcmsExperiment'
featureChromatograms(
object,
expandRt = 0,
expandMz = 0,
aggregationFun = "max",
features = character(),
return.type = "XChromatograms",
chunkSize = 2L,
mzmin = min,
mzmax = max,
rtmin = min,
rtmax = max,
...,
progressbar = TRUE,
BPPARAM = bpparam()
)
# S4 method for class 'XCMSnExp'
featureChromatograms(
object,
expandRt = 0,
aggregationFun = "max",
features,
include = c("feature_only", "apex_within", "any", "all"),
filled = FALSE,
n = length(fileNames(object)),
value = c("maxo", "into"),
expandMz = 0,
...
)Arguments
- object
XcmsExperimentorXCMSnExpobject with grouped chromatographic peaks.- ...
optional arguments to be passed along to the
chromatogram()function.- expandRt
numeric(1)to expand the retention time range for each chromatographic peak by a constant value on each side.- expandMz
numeric(1)to expand the m/z range for each chromatographic peak by a constant value on each side. Be aware that by extending the m/z range the extracted EIC might no longer represent the actual identified chromatographic peak because intensities of potential additional mass peaks within each spectra would be aggregated into the final reported intensity value per spectrum (retention time).- aggregationFun
character(1)specifying the name that should be used to aggregate intensity values across the m/z value range for the same retention time. The default"max"returns a base peak chromatogram.- features
integer,characterorlogicaldefining a subset of features for which chromatograms should be returned. Can be the index of the features infeatureDefinitions, feature IDs (row names offeatureDefinitions) or a logical vector.- return.type
character(1)defining how the result should be returned. At present onlyreturn.type = "XChromatograms"is supported and the results are thus returned as anXChromatograms()object.- chunkSize
For
objectbeing anXcmsExperiment:integer(1)defining the number of files from which the data should be loaded at a time into memory. Defaults tochunkSize = 2L.- mzmin
functiondefining how the lower boundary of the m/z region from which the EIC is integrated should be defined. Defaults tomzmin = minthus the smallest"mzmin"value for all chromatographic peaks of a feature will be used.- mzmax
functiondefining how the upper boundary of the m/z region from which the EIC is integrated should be defined. Defaults tomzmax = maxthus the largest"mzmax"value for all chromatographic peaks of a feature will be used.- rtmin
functiondefining how the lower boundary of the rt region from which the EIC is integrated should be defined. Defaults tortmin = minthus the smallest"rtmin"value for all chromatographic peaks of a feature will be used.- rtmax
functiondefining how the upper boundary of the rt region from which the EIC is integrated should be defined. Defaults tortmax = maxthus the largest"rtmax"value for all chromatographic peaks of a feature will be used.- progressbar
logical(1)defining whether a progress bar is shown.- BPPARAM
For
objectbeing anXcmsExperiment: parallel processing setup. Defaults toBPPARAM = bpparam(). SeeBiocParallel::bpparam()for more information.- include
Only for
objectbeing anXCMSnExp:character(1)defining which chromatographic peaks (and related feature definitions) should be included in the returnedXChromatograms(). Defaults to"feature_only"; See description above for options and details.- filled
Only for
objectbeing anXCMSnExp:logical(1)whether filled-in peaks should be included in the result object. The default isfilled = FALSE, i.e. only detected peaks are reported.- n
Only for
objectbeing anXCMSnExp:integer(1)to optionally specify the number of top n samples from which the EIC should be extracted.- value
Only for
objectbeing anXCMSnExp:character(1)specifying the column to be used to sort the samples. Can be either"maxo"(the default) or"into"to use the maximal peak intensity or the integrated peak area, respectively.
Value
XChromatograms() object. In future, depending on parameter
return.type, the data might be returned as a different object.
Note
The EIC data of a feature is extracted from every sample using the same
m/z - rt area. The EIC in a sample does thus not exactly represent the
signal of the actually identified chromatographic peak in that sample.
The chromPeakChromatograms() function would allow to extract the actual
EIC of the chromatographic peak in a specific sample. See also examples
below.
Parameters include, filled, n and value are only supported
for object being an XCMSnExp.
When extracting EICs from only the top n samples it can happen that one
or more of the features specified with features are dropped because they
have no detected peak in the top n samples. The chance for this to happen
is smaller if x contains also filled-in peaks (with fillChromPeaks).
See also
filterColumnsKeepTop() to filter the extracted EICs keeping only
the top n columns (samples) with the highest intensity.
chromPeakChromatograms() for a function to extract an EIC for each
chromatographic peak.
Examples
## Load a test data set with detected peaks
library(xcms)
library(MsExperiment)
faahko_sub <- loadXcmsData("faahko_sub2")
## Disable parallel processing for this example
register(SerialParam())
## Perform correspondence analysis
xdata <- groupChromPeaks(faahko_sub,
param = PeakDensityParam(minFraction = 0.8, sampleGroups = rep(1, 3)))
## Get the feature definitions
featureDefinitions(xdata)
#> mzmed mzmin mzmax rtmed rtmin rtmax npeaks 1 peakidx
#> FT01 300.2 300.2 300.2 3387.143 3379.317 3390.271 4 3 35, 125,....
#> FT02 301.0 301.0 301.0 2787.766 2786.200 2792.459 3 3 10, 97, 198
#> FT03 326.2 326.2 326.2 3416.877 3415.311 3424.700 3 3 37, 128, 213
#> FT04 328.2 328.2 328.2 3626.581 3618.755 3631.274 3 3 64, 159, 231
#> FT05 329.2 329.2 329.2 3626.581 3618.755 3631.274 3 3 63, 156, 230
#> FT06 343.0 343.0 343.0 2684.479 2678.218 2686.042 3 3 6, 92, 195
#> FT07 344.0 344.0 344.0 2682.914 2679.783 2686.042 3 3 3, 90, 193
#> FT08 354.2 354.2 354.2 3614.061 3610.930 3621.884 3 3 56, 153, 228
#> FT09 356.2 356.2 356.2 3830.025 3819.069 3833.153 3 3 78, 180, 241
#> FT10 365.0 365.0 365.0 2684.479 2679.783 2686.042 3 3 7, 93, 196
#> FT11 438.3 438.3 438.3 4052.248 4047.552 4060.071 3 3 81, 186, 248
#> FT12 496.2 496.2 496.2 3335.498 3316.719 3340.194 3 3 47, 143, 220
#> FT13 496.2 496.2 496.2 3402.791 3384.012 3410.617 3 3 48, 144, 221
#> FT14 508.2 508.2 508.2 3524.859 3515.468 3529.552 3 3 67, 163, 232
#> FT15 509.2 509.2 509.2 3523.294 3512.338 3531.117 3 3 54, 160, 229
#> FT16 522.2 522.2 522.2 3387.142 3344.888 3434.091 6 3 52, 53, ....
#> FT17 523.2 523.2 523.2 3387.142 3344.888 3434.091 6 3 38, 39, ....
#> FT18 524.2 524.2 524.2 3676.658 3598.410 3704.828 4 3 74, 75, ....
#> FT19 525.2 525.2 525.2 3690.742 3662.574 3706.393 3 3 72, 173, 238
#> FT20 532.2 532.2 532.2 3482.605 3476.344 3488.863 3 3 49, 137, 219
#> FT21 536.2 536.2 536.2 3712.653 3703.262 3718.912 3 3 73, 174, 237
#> ms_level
#> FT01 1
#> FT02 1
#> FT03 1
#> FT04 1
#> FT05 1
#> FT06 1
#> FT07 1
#> FT08 1
#> FT09 1
#> FT10 1
#> FT11 1
#> FT12 1
#> FT13 1
#> FT14 1
#> FT15 1
#> FT16 1
#> FT17 1
#> FT18 1
#> FT19 1
#> FT20 1
#> FT21 1
## Extract ion chromatograms for the first 3 features. Parameter
## `features` can be either the feature IDs or feature indices.
chrs <- featureChromatograms(xdata,
features = rownames(featureDefinitions)[1:3])
#> Processing chromatographic peaks for features
## Plot the EIC for the first feature using different colors for each file.
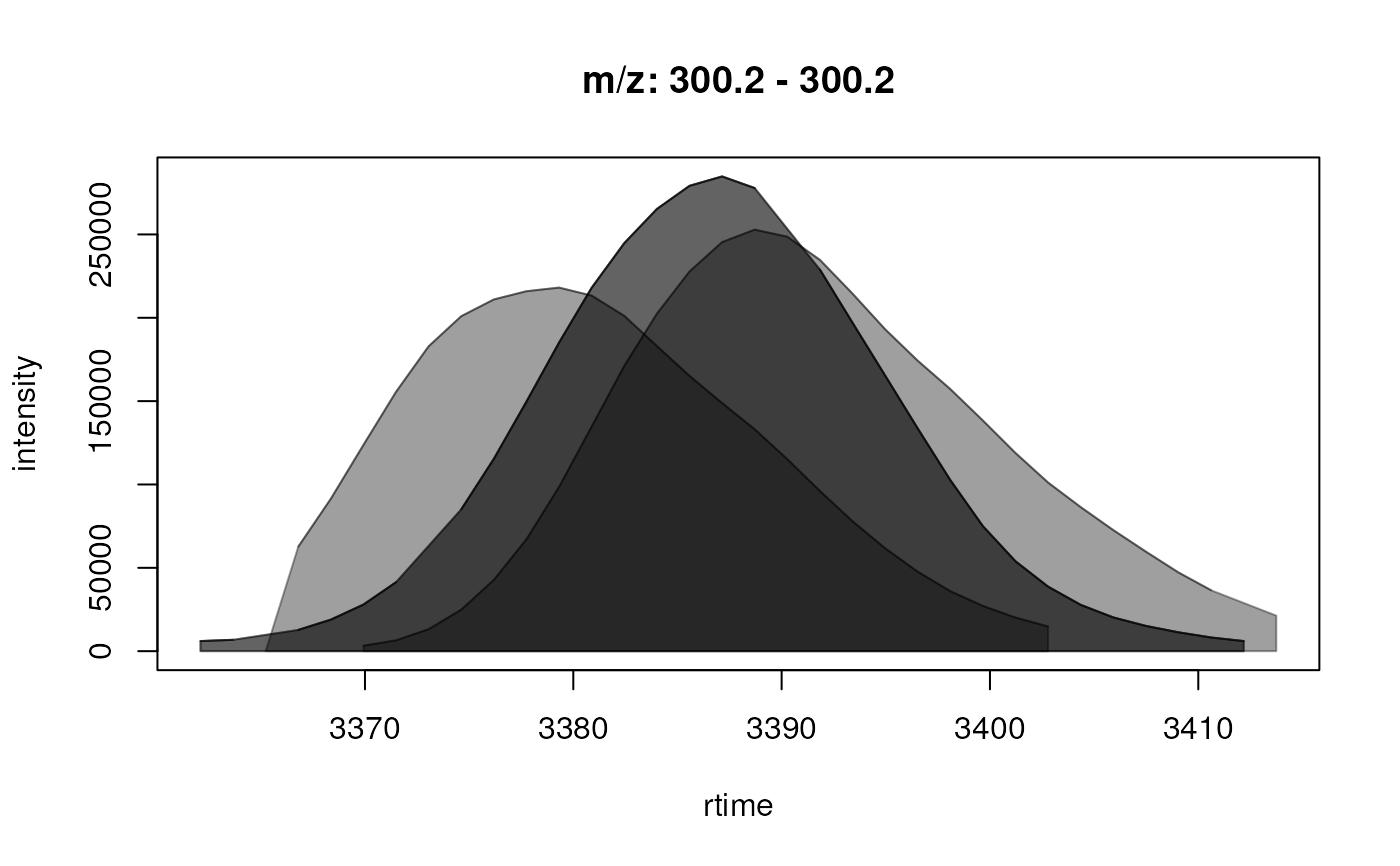
plot(chrs[1, ], col = c("red", "green", "blue"))
 ## The EICs for all 3 samples use the same m/z and retention time range,
## which was defined using the `featureArea` function:
featureArea(xdata, features = rownames(featureDefinitions(xdata))[1:3],
mzmin = min, mzmax = max, rtmin = min, rtmax = max)
#> mzmin mzmax rtmin rtmax
#> FT01 300.2 300.2 3362.104 3413.746
#> FT02 301.0 301.0 2765.855 2820.630
#> FT03 326.2 326.2 3391.837 3449.740
## To extract the actual (exact) EICs for each chromatographic peak of
## a feature in each sample, the `chromPeakChromatograms` function would
## need to be used instead. Below we extract the EICs for all
## chromatographic peaks of the first feature. We need to first get the
## IDs of all chromatographic peaks assigned to the first feature:
peak_ids <- rownames(chromPeaks(xdata))[featureDefinitions(xdata)$peakidx[[1L]]]
## We can now pass these to the `chromPeakChromatograms` function with
## parameter `peaks`:
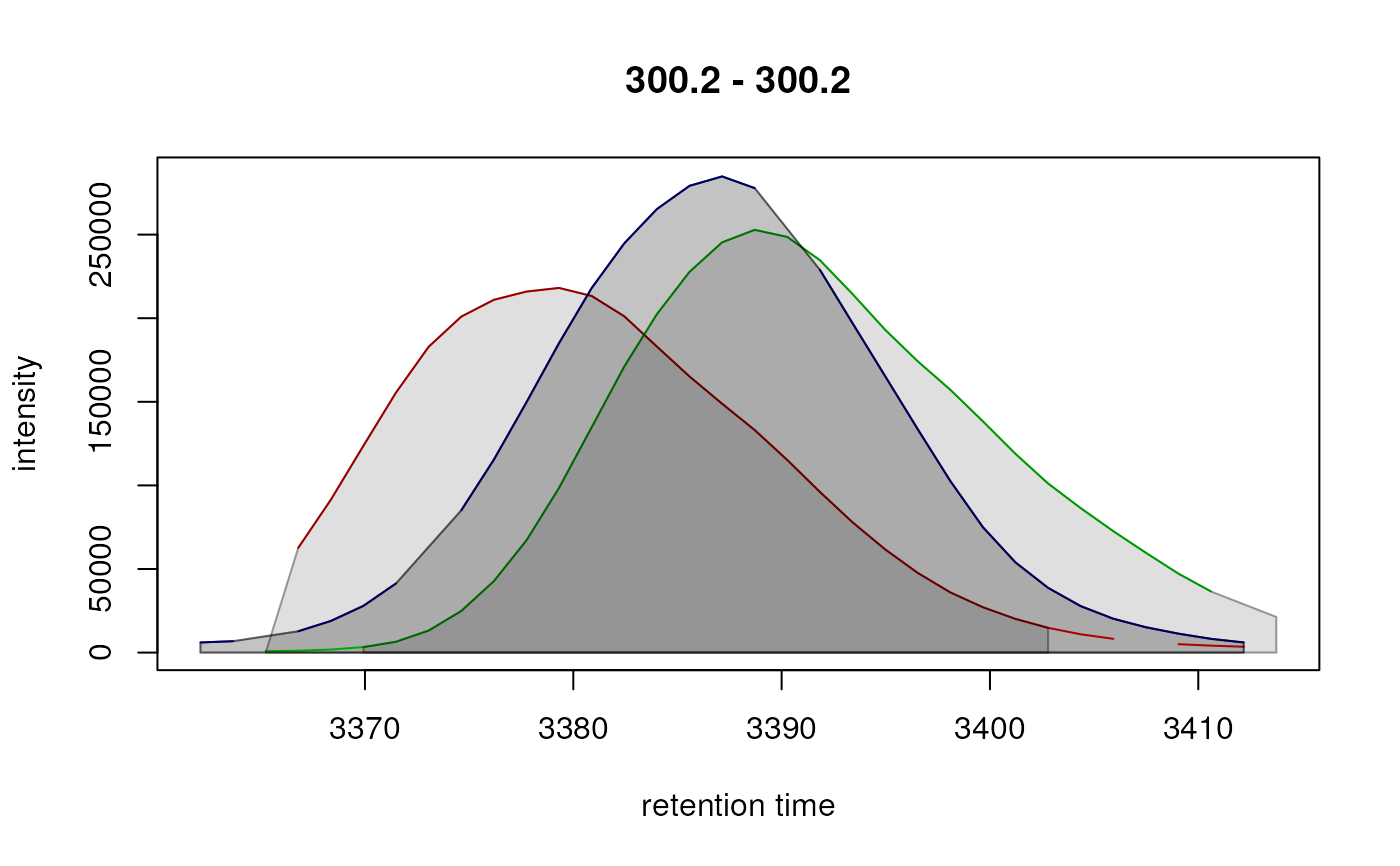
eic_1 <- chromPeakChromatograms(xdata, peaks = peak_ids)
## To plot these into a single plot we need to use the
## `plotChromatogramsOverlay` function:
plotChromatogramsOverlay(eic_1)
## The EICs for all 3 samples use the same m/z and retention time range,
## which was defined using the `featureArea` function:
featureArea(xdata, features = rownames(featureDefinitions(xdata))[1:3],
mzmin = min, mzmax = max, rtmin = min, rtmax = max)
#> mzmin mzmax rtmin rtmax
#> FT01 300.2 300.2 3362.104 3413.746
#> FT02 301.0 301.0 2765.855 2820.630
#> FT03 326.2 326.2 3391.837 3449.740
## To extract the actual (exact) EICs for each chromatographic peak of
## a feature in each sample, the `chromPeakChromatograms` function would
## need to be used instead. Below we extract the EICs for all
## chromatographic peaks of the first feature. We need to first get the
## IDs of all chromatographic peaks assigned to the first feature:
peak_ids <- rownames(chromPeaks(xdata))[featureDefinitions(xdata)$peakidx[[1L]]]
## We can now pass these to the `chromPeakChromatograms` function with
## parameter `peaks`:
eic_1 <- chromPeakChromatograms(xdata, peaks = peak_ids)
## To plot these into a single plot we need to use the
## `plotChromatogramsOverlay` function:
plotChromatogramsOverlay(eic_1)