Grouping FTICR-MS data with xcms
Joachim Bargsten
Johannes Rainer
Source:vignettes/xcms-direct-injection.Rmd
xcms-direct-injection.RmdIntroduction
This document describes how to use xcms for the analysis of direct injection mass spec data, including peak detection, calibration and correspondence (grouping of peaks across samples).
Peak detection
Prior to any other analysis step, peaks have to be identified in the mass spec data. In contrast to the typical metabolomics workflow, in which peaks are identified in the chromatographic (time) dimension, in direct injection mass spec data sets peaks are identified in the m/z dimension. xcms uses functionality from the MassSpecWavelet package to identify such peaks.
Below we load the required packages. For information on the parallel processing setup please see the BiocParallel vignette.
In this documentation we use an example data set from the
r Biocpkg("MsDataHub") package. Assuming that MsDataHub
is installed, it will obtain the files from https://doi.org/10.5281/zenodo.18494293 and load the
data set. We create also a data.frame describing the
experimental setup based on the file names.
## We're using 2 samples per condition
## from https://doi.org/10.5281/zenodo.18494293
mzML_files <- c(
HAM004_641fE_14.11.07..Exp1.extracted.mzML(),
HAM004_641fE_14.11.07..Exp2.extracted.mzML(),
HAM005_641fE_14.11.07..Exp1.extracted.mzML(),
HAM005_641fE_14.11.07..Exp2.extracted.mzML()
)
## Create a data.frame assigning samples to sample groups, i.e. ham4 and ham5.
grp <- c("ham4", "ham4", "ham5", "ham5")
pd <- data.frame(filename = basename(mzML_files), sample_group = grp)
## Load the data.
ham_raw <- readMSData(files = mzML_files,
pdata = AnnotatedDataFrame(pd),
mode = "onDisk")## Warning in .testReadMSDataInput(environment()): Reading different file formats in.
## This is untested and you are welcome to try it out.
## Please report back!The data files are from direct injection mass spectrometry experiments, i.e. we have only a single spectrum available for each sample and no retention times.
## Only a single spectrum with an *artificial* retention time is available
## for each sample
rtime(ham_raw)## F1.S1 F2.S1 F3.S1 F4.S1
## -1 -1 -1 -1Peaks are identified within each spectrum using the mass spec wavelet method.
## Define the parameters for the peak detection
msw <- MSWParam(scales = c(1, 4, 9), nearbyPeak = TRUE, winSize.noise = 500,
SNR.method = "data.mean", snthresh = 10)
ham_prep <- findChromPeaks(ham_raw, param = msw)
head(chromPeaks(ham_prep))## mz mzmin mzmax rt rtmin rtmax into maxo sn intf
## CP01 403.2367 403.2279 403.2447 -1 -1 -1 4735258 372259.4 22.97534 NA
## CP02 409.1845 409.1747 409.1936 -1 -1 -1 4158404 310572.1 20.61382 NA
## CP03 413.2677 413.2585 413.2769 -1 -1 -1 6099006 435462.6 27.21723 NA
## CP04 423.2363 423.2266 423.2459 -1 -1 -1 2708391 174252.7 14.74527 NA
## CP05 427.2681 427.2574 427.2779 -1 -1 -1 6302089 461385.6 32.50050 NA
## CP06 437.2375 437.2254 437.2488 -1 -1 -1 7523070 517917.6 34.37645 NA
## maxf sample
## CP01 814693.1 1
## CP02 732119.9 1
## CP03 1018994.8 1
## CP04 435858.5 1
## CP05 1125644.3 1
## CP06 1282906.5 1Calibration
The calibrate method can be used to correct the m/z
values of identified peaks. The currently implemented method requires
identified peaks and a list of m/z values for known calibrants. The
identified peaks m/z values are then adjusted based on the differences
between the calibrants’ m/z values and the m/z values of the closest
peaks (within a user defined permitted maximal distance). Note that this
method does presently only calibrate identified peaks, but not the
original m/z values in the spectra.
Below we demonstrate the calibrate method on one of the
data files with artificially defined calibration m/z values. We first
subset the data set to the first data file, extract the m/z values of 3
peaks and modify the values slightly.
## Subset to the first file.
first_file <- filterFile(ham_prep, file = 1)
## Extract 3 m/z values
calib_mz <- chromPeaks(first_file)[c(1, 4, 7), "mz"]
calib_mz <- calib_mz + 0.00001 * runif(1, 0, 0.4) * calib_mz + 0.0001Next we calibrate the data set using the previously defined
artificial calibrants. We are using the
"edgeshift" method for calibration that adjusts all peaks
within the range of the m/z values of the calibrants using a linear
interpolation and shifts all chromatographic peaks outside of that range
by a constant factor (the difference between the lowest respectively
largest calibrant m/z with the closest peak’s m/z). Note that in a
real use case, the m/z values would obviously represent known
m/z of calibrants and would not be defined on the actual data.
## Set-up the parameter class for the calibration
prm <- CalibrantMassParam(mz = calib_mz, method = "edgeshift",
mzabs = 0.0001, mzppm = 5)
first_file_calibrated <- calibrate(first_file, param = prm)To evaluate the calibration we plot below the difference between the adjusted and raw m/z values (y-axis) against the raw m/z values.
diffs <- chromPeaks(first_file_calibrated)[, "mz"] -
chromPeaks(first_file)[, "mz"]
plot(x = chromPeaks(first_file)[, "mz"], xlab = expression(m/z[raw]),
y = diffs, ylab = expression(m/z[calibrated] - m/z[raw]))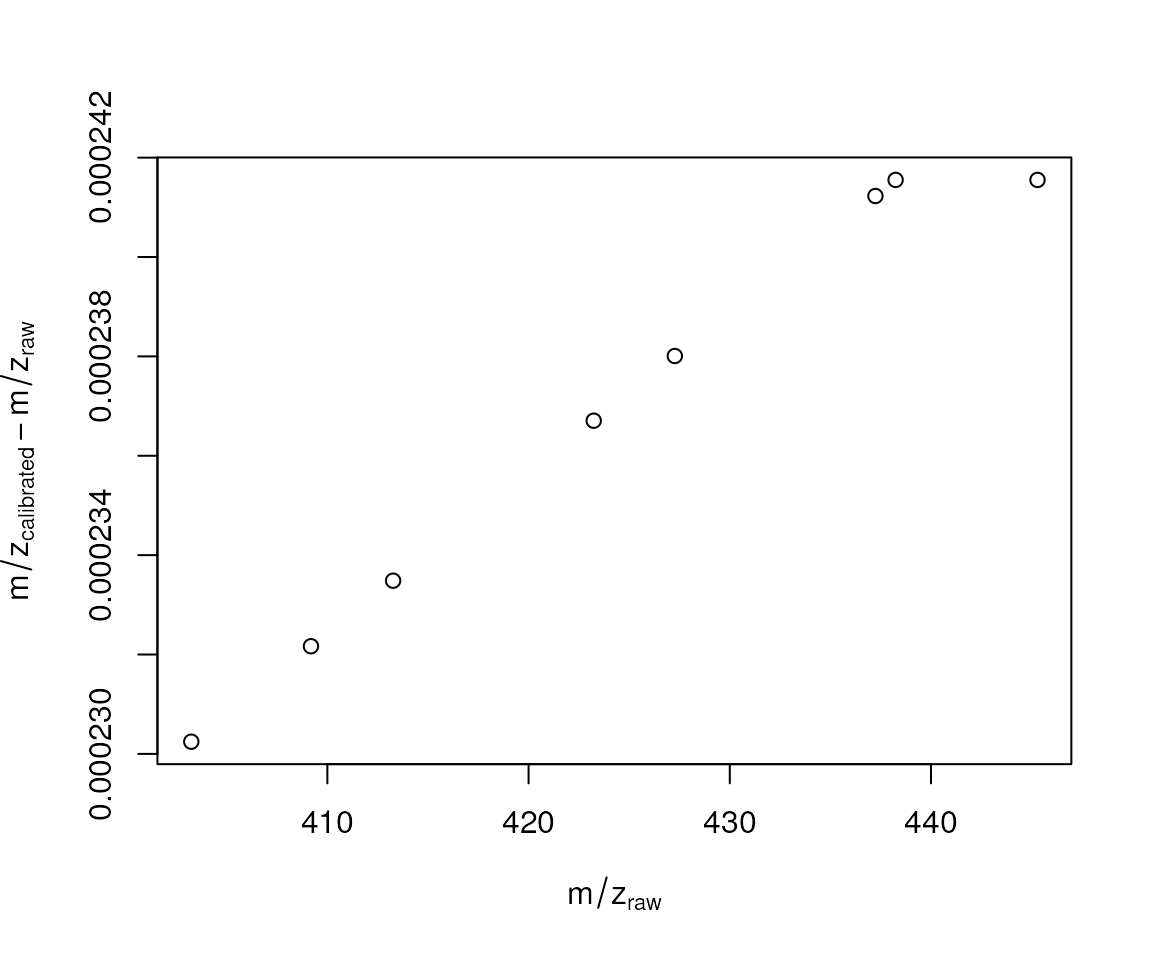
Correspondence
Correspondence aims to group peaks across samples to define the
features (ions with the same m/z values). Peaks from single
spectrum, direct injection MS experiments can be grouped with the
MZclust method. Below we perform the correspondence analysis
with the groupChromPeaks method using default settings.
## Using default settings but define sample group assignment
mzc_prm <- MzClustParam(sampleGroups = ham_prep$sample_group)
ham_prep <- groupChromPeaks(ham_prep, param = mzc_prm)Getting an overview of the performed processings:
ham_prep## MSn experiment data ("XCMSnExp")
## Object size in memory: 0.04 Mb
## - - - Spectra data - - -
## MS level(s): 1
## Number of spectra: 4
## MSn retention times: -1:59 - -1:59 minutes
## - - - Processing information - - -
## Data loaded [Sun Jul 5 13:03:50 2026]
## MSnbase version: 2.37.0
## - - - Meta data - - -
## phenoData
## rowNames: 1 2 3 4
## varLabels: filename sample_group
## varMetadata: labelDescription
## Loaded from:
## [1] 3be765196f0_10386... [4] 3be7c059423_10392
## Use 'fileNames(.)' to see all files.
## protocolData: none
## featureData
## featureNames: F1.S1 F2.S1 F3.S1 F4.S1
## fvarLabels: fileIdx spIdx ... spectrum (36 total)
## fvarMetadata: labelDescription
## experimentData: use 'experimentData(object)'
## - - - xcms preprocessing - - -
## Chromatographic peak detection:
## method: MSW
## 38 peaks identified in 4 samples.
## On average 9.5 chromatographic peaks per sample.
## Correspondence:
## method: mzClust
## 20 features identified.
## Median mz range of features: 9.1553e-05
## Median rt range of features: 0The peak group information, i.e. the feature definitions can
be accessed with the featureDefinitions method.
featureDefinitions(ham_prep)## DataFrame with 20 rows and 10 columns
## mzmed mzmin mzmax rtmed rtmin rtmax npeaks
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## FT01 402.285 402.285 402.286 -1 -1 -1 2
## FT02 403.237 403.237 403.237 -1 -1 -1 4
## FT03 405.109 405.109 405.109 -1 -1 -1 2
## FT04 409.184 409.184 409.185 -1 -1 -1 2
## FT05 410.144 410.144 410.145 -1 -1 -1 2
## ... ... ... ... ... ... ... ...
## FT16 437.238 437.238 437.238 -1 -1 -1 2
## FT17 438.240 438.240 438.240 -1 -1 -1 2
## FT18 439.151 439.151 439.151 -1 -1 -1 2
## FT19 441.130 441.130 441.131 -1 -1 -1 2
## FT20 445.293 445.292 445.293 -1 -1 -1 2
## ham4 ham5 peakidx
## <numeric> <numeric> <list>
## FT01 0 2 16,28
## FT02 2 2 17,29,1,...
## FT03 0 2 18,30
## FT04 2 0 10,2
## FT05 0 2 19,31
## ... ... ... ...
## FT16 2 0 6,13
## FT17 2 0 7,14
## FT18 0 2 26,37
## FT19 0 2 38,27
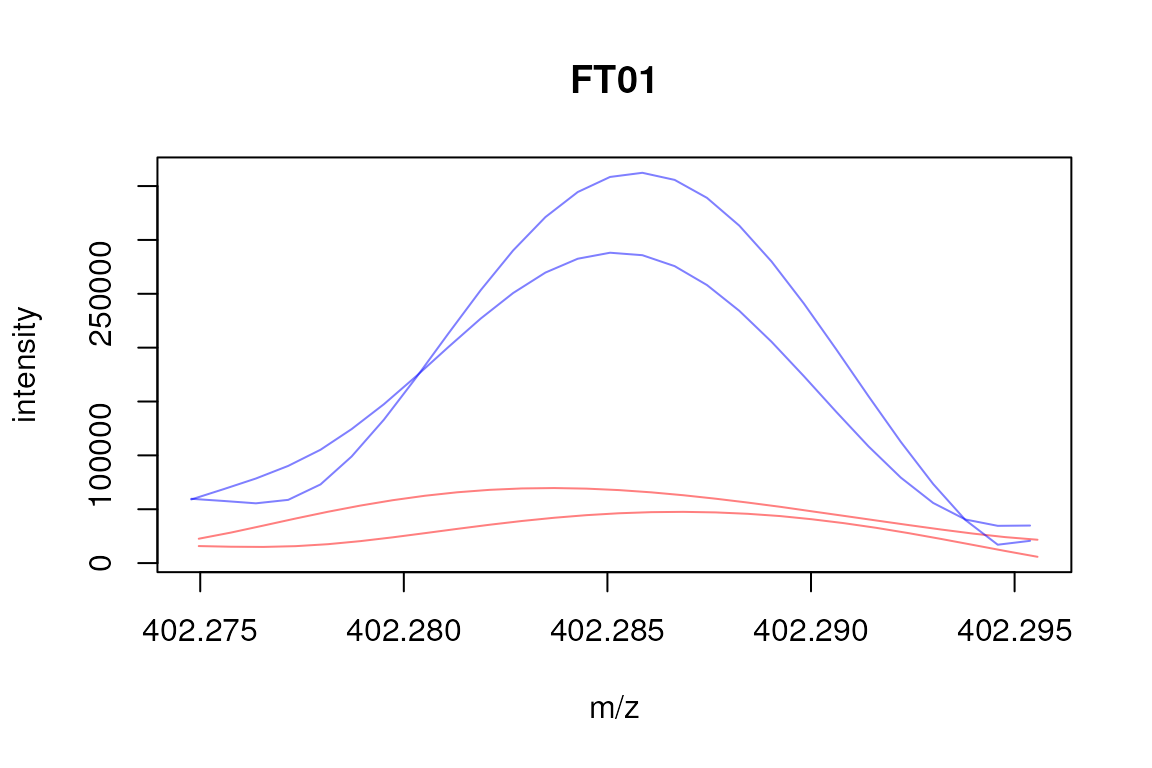
## FT20 2 0 15,8Plotting the raw data for direct injection samples involves a little
more processing than for LC/GC-MS data in which we can simply use the
chromatogram method to extract the data. Below we extract
the m/z-intensity pairs for the peaks associated with the first feature.
We thus first identify the peaks for that feature and define their m/z
values range. Using this range we can subsequently use the
filterMz function to sub-set the full data set to the
signal associated with the feature’s peaks. On that object we can then
call the mz and intensity functions to extract
the data.
## Get the peaks belonging to the first feature
pks <- chromPeaks(ham_prep)[featureDefinitions(ham_prep)$peakidx[[1]], ]
## Define the m/z range
mzr <- c(min(pks[, "mzmin"]) - 0.001, max(pks[, "mzmax"]) + 0.001)
## Subset the object to the m/z range
ham_prep_sub <- filterMz(ham_prep, mz = mzr)
## Extract the mz and intensity values
mzs <- mz(ham_prep_sub, bySample = TRUE)
ints <- intensity(ham_prep_sub, bySample = TRUE)
## Plot the data
plot(3, 3, pch = NA, xlim = range(mzs), ylim = range(ints), main = "FT01",
xlab = "m/z", ylab = "intensity")
## Define colors
cols <- rep("#ff000080", length(mzs))
cols[ham_prep_sub$sample_group == "ham5"] <- "#0000ff80"
tmp <- mapply(mzs, ints, cols, FUN = function(x, y, col) {
points(x, y, col = col, type = "l")
})
To access the actual intensity values of each feature in each sample
the featureValue method can be used. The setting
value = "into" tells the function to return the integrated
signal for each peak (one representative peak) per sample.
feat_vals <- featureValues(ham_prep, value = "into")
head(feat_vals)## 3be765196f0_10386 3be72090e774_10387 3be73bdf6cb9_10391 3be7c059423_10392
## FT01 NA NA 4095293 4804763
## FT02 4735258 6202418 4811391 2581183
## FT03 NA NA 2982453 2268984
## FT04 4158404 5004546 NA NA
## FT05 NA NA 2872023 2133219
## FT06 6099006 4950642 NA NANA is reported for features in samples for which no peak
was identified at the feature’s m/z value. In some instances there might
still be a signal at the feature’s position in the raw data files, but
the peak detection failed to identify a peak. For these cases signal can
be recovered using the fillChromPeaks method that
integrates all raw signal at the feature’s location. If there is no
signal at that location an NA is reported.
ham_prep <- fillChromPeaks(ham_prep, param = FillChromPeaksParam())
head(featureValues(ham_prep, value = "into"))## 3be765196f0_10386 3be72090e774_10387 3be73bdf6cb9_10391 3be7c059423_10392
## FT01 768754.0 1230140.4 4095293 4804762.5
## FT02 4735257.5 6202417.6 4811391 2581183.1
## FT03 652566.6 374109.9 2982453 2268984.5
## FT04 4158404.5 5004546.3 1221031 1241294.4
## FT05 652201.1 403448.4 2872023 2133219.4
## FT06 6099006.3 4950641.7 1573988 977694.5Further analysis
Further analysis, i.e. detection of features/metabolites with significantly different abundances, or PCA analyses can be performed on the feature matrix using functionality from other R packages, such as limma.
Session information
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] MsDataHub_1.12.0 MassSpecWavelet_1.78.0 xcms_4.11.1
## [4] BiocParallel_1.46.0 MSnbase_2.37.0 ProtGenerics_1.44.0
## [7] S4Vectors_0.50.1 mzR_2.46.0 Rcpp_1.1.1-1.1
## [10] Biobase_2.72.0 BiocGenerics_0.58.1 generics_0.1.4
## [13] BiocStyle_2.40.0
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0
## [3] MultiAssayExperiment_1.38.0 magrittr_2.0.5
## [5] farver_2.1.2 MALDIquant_1.22.3
## [7] rmarkdown_2.31 fs_2.1.0
## [9] ragg_1.5.2 vctrs_0.7.3
## [11] memoise_2.0.1 htmltools_0.5.9
## [13] S4Arrays_1.12.0 progress_1.2.3
## [15] AnnotationHub_4.2.2 curl_7.1.0
## [17] signal_1.8-1 SparseArray_1.12.2
## [19] mzID_1.50.0 sass_0.4.10
## [21] bslib_0.11.0 htmlwidgets_1.6.4
## [23] desc_1.4.3 plyr_1.8.9
## [25] httr2_1.2.2 impute_1.86.0
## [27] cachem_1.1.0 igraph_2.3.2
## [29] lifecycle_1.0.5 iterators_1.0.14
## [31] pkgconfig_2.0.3 Matrix_1.7-5
## [33] R6_2.6.1 fastmap_1.2.0
## [35] MatrixGenerics_1.24.0 clue_0.3-68
## [37] digest_0.6.39 pcaMethods_2.4.0
## [39] AnnotationDbi_1.74.0 ExperimentHub_3.2.0
## [41] textshaping_1.0.5 GenomicRanges_1.64.0
## [43] RSQLite_3.53.2 filelock_1.0.3
## [45] Spectra_1.22.2 httr_1.4.8
## [47] abind_1.4-8 compiler_4.6.0
## [49] withr_3.0.3 bit64_4.8.2
## [51] doParallel_1.0.17 S7_0.2.2
## [53] PTMods_1.0.0 DBI_1.3.0
## [55] MASS_7.3-65 MsExperiment_1.14.0
## [57] rappdirs_0.3.4 DelayedArray_0.38.2
## [59] tools_4.6.0 PSMatch_1.16.0
## [61] otel_0.2.0 glue_1.8.1
## [63] QFeatures_1.22.0 grid_4.6.0
## [65] cluster_2.1.8.2 reshape2_1.4.5
## [67] gtable_0.3.6 preprocessCore_1.74.0
## [69] tidyr_1.3.2 data.table_1.18.4
## [71] hms_1.1.4 MetaboCoreUtils_1.20.1
## [73] XVector_0.52.0 BiocVersion_3.23.1
## [75] foreach_1.5.2 pillar_1.11.1
## [77] stringr_1.6.0 limma_3.68.4
## [79] dplyr_1.2.1 BiocFileCache_3.2.0
## [81] lattice_0.22-9 bit_4.6.0
## [83] tidyselect_1.2.1 Biostrings_2.80.1
## [85] knitr_1.51 bookdown_0.47
## [87] IRanges_2.46.0 Seqinfo_1.2.0
## [89] SummarizedExperiment_1.42.0 xfun_0.59
## [91] statmod_1.5.2 matrixStats_1.5.0
## [93] stringi_1.8.7 lazyeval_0.2.3
## [95] yaml_2.3.12 evaluate_1.0.5
## [97] codetools_0.2-20 MsCoreUtils_1.24.0
## [99] tibble_3.3.1 BiocManager_1.30.27
## [101] cli_3.6.6 affyio_1.82.0
## [103] systemfonts_1.3.2 jquerylib_0.1.4
## [105] dbplyr_2.6.0 png_0.1-9
## [107] XML_3.99-0.23 parallel_4.6.0
## [109] pkgdown_2.2.0.9000 ggplot2_4.0.3
## [111] blob_1.3.0 prettyunits_1.2.0
## [113] AnnotationFilter_1.36.0 MsFeatures_1.20.0
## [115] scales_1.4.0 affy_1.90.0
## [117] ncdf4_1.24 purrr_1.2.2
## [119] crayon_1.5.3 rlang_1.2.0
## [121] vsn_3.80.0 KEGGREST_1.52.2