Package: xcms
Authors: Johannes Rainer, Michael Witting
Modified: 2026-07-05 12:02:17.561418
Compiled: Sun Jul 5 13:03:59 2026
Introduction
Metabolite identification is an important step in non-targeted metabolomics and requires different steps. One involves the use of tandem mass spectrometry to generate fragmentation spectra of detected metabolites (LC-MS/MS), which are then compared to fragmentation spectra of known metabolites. Different approaches exist for the generation of these fragmentation spectra, whereas the most used is data dependent acquisition (DDA) also known as the top-n method. In this method the top N most intense ions (m/z values) from a MS1 scan are selected for fragmentation in the next N scans before the cycle starts again. This method allows to generate clean MS2 fragmentation spectra on the fly during acquisition without the need for further experiments, but suffers from poor coverage of the detected metabolites (since only a limited number of ions are fragmented) and less accurate quantification of the compounds (since fewer MS1 scans are generated).
Data independent approaches (DIA) like Bruker bbCID, Agilent AllIons or Waters MSe don’t use such a preselection, but rather fragment all detected molecules at once. They are using alternating schemes with scan of low and high collision energy to collect MS1 and MS2 data. Using this approach, there is no problem in coverage, but the relation between the precursor and fragment masses is lost leading to chimeric spectra. Sequential Window Acquisition of all Theoretical Mass Spectra (or SWATH [1]) combines both approaches through a middle-way approach. There is no precursor selection and acquisition is independent of acquired data, but rather than isolating all precusors at once, defined windows (i.e. ranges of m/z values) are used and scanned. This reduces the overlap of fragment spectra while still keeping a high coverage.
This document showcases the analysis of two small LC-MS/MS data sets using xcms. The data files used are reversed-phase LC-MS/MS runs from the Agilent Pesticide mix obtained from a Sciex 6600 Triple ToF operated in SWATH acquisition mode. For comparison a DDA file from the same sample is included.
See also the Metabonaut resource for other examples of LC-MS/MS analysis workflows with xcms.
Analysis of DDA data
Below we load the example DDA data set and create a total ion chromatogram of its MS1 data.
library(xcms)
library(MsExperiment)
library(MsDataHub)
dda_file <- PestMix1_DDA.mzML()
dda_data <- readMsExperiment(dda_file)
chr <- chromatogram(dda_data, aggregationFun = "sum", msLevel = 1L)According to the TIC most of the signal is measured between ~ 200 and 600 seconds (see plot below). We thus filter the DDA data to this retention time range.
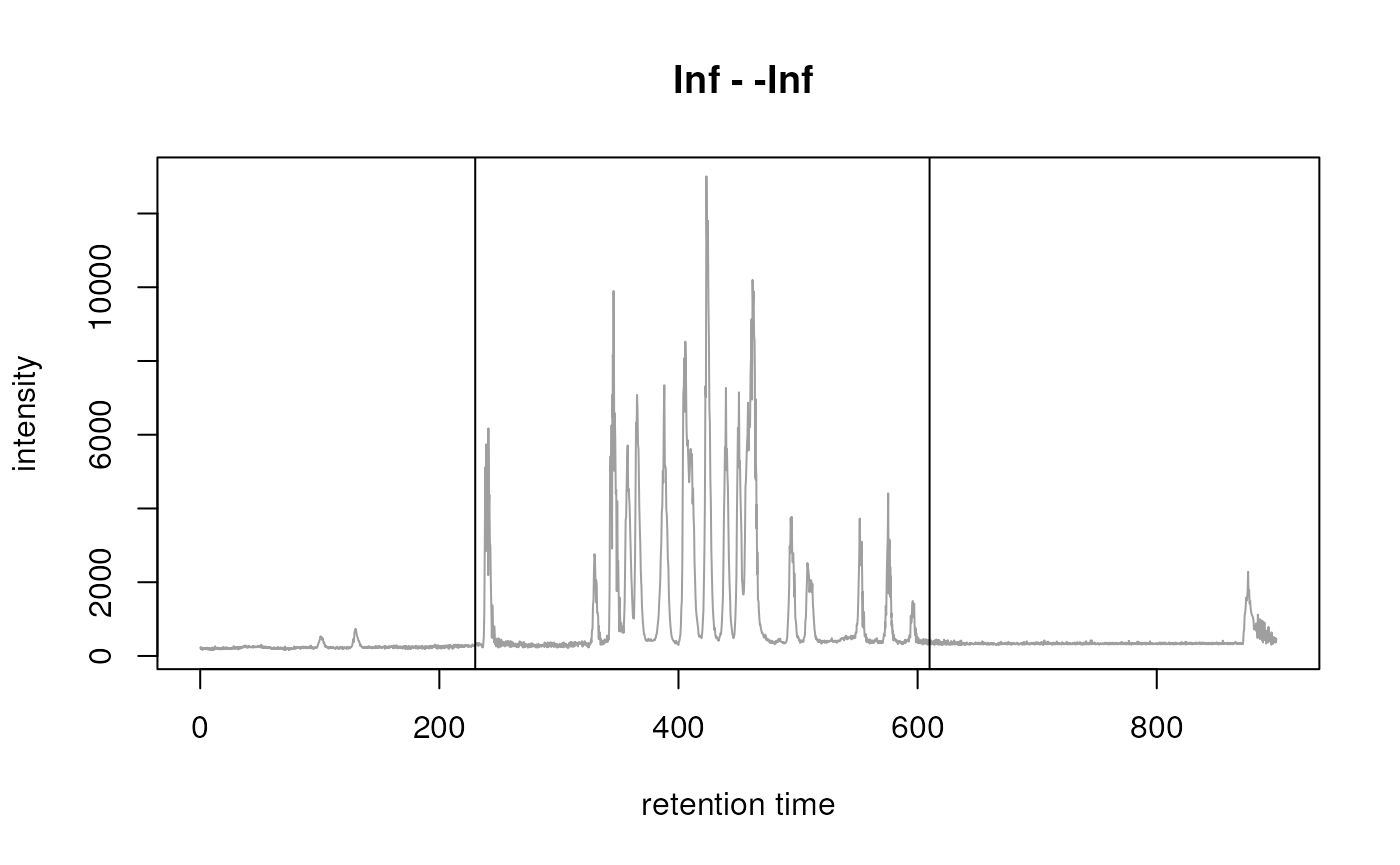
## Filter spectraThe variable dda_data contains now all MS1 and MS2
spectra from the specified mzML file. The number of spectra for each MS
level is listed below. Note that we subset the experiment to the first
data file (using [1]) and then access directly the spectra
within this sample with the spectra() function (which
returns a Spectra object from the Spectra
package). Note that we use the pipe operator |> for
better readability.
##
## 1 2
## 1389 2214For the MS2 spectra we can get the m/z of the precursor ion with the
precursorMz() function. Below we first subset the data set
again to a single sample and filter to spectra from MS level 2
extracting then their precursor m/z values.
dda_data[1] |>
spectra() |>
filterMsLevel(2) |>
precursorMz() |>
head()## [1] 182.18777 182.18893 55.00579 182.19032 237.12296 237.11987With the precursorIntensity() function it is also
possible to extract the intensity of the precursor ion.
dda_data[1] |>
spectra() |>
filterMsLevel(2) |>
precursorIntensity() |>
head()## [1] 0 0 0 0 0 0Some manufacturers (like Sciex) don’t define/export the precursor
intensity and thus either NA or 0 is reported.
We can however use the estimatePrecursorIntensity()
function from the Spectra
package to determine the precursor intensity for a MS 2 spectrum based
on the intensity of the respective ion in the previous MS1 scan (note
that with method = "interpolation" the precursor intensity
would be defined based on interpolation between the intensity in the
previous and subsequent MS1 scan). Below we estimate the precursor
intensities, on the full data (for MS1 spectra a NA value
is reported).
prec_int <- estimatePrecursorIntensity(spectra(dda_data))We next set the precursor intensity in the spectrum metadata of
dda_data. So that it can be extracted later with the
precursorIntensity() function.
spectra(dda_data)$precursorIntensity <- prec_int
dda_data[1] |>
spectra() |>
filterMsLevel(2) |>
precursorIntensity() |>
head()## [1] NA 9.198155 2.773988 27.590797 3.443145 7.621923Next we perform the chromatographic peak detection on the MS level 1
data with the findChromPeaks() method. Below we define the
settings for a centWave-based peak detection and perform the
analysis.
cwp <- CentWaveParam(snthresh = 5, noise = 100, ppm = 10,
peakwidth = c(3, 30))
dda_data <- findChromPeaks(dda_data, param = cwp, msLevel = 1L)In total 114 peaks were identified in the present data set.
The advantage of LC-MS/MS data is that (MS1) ions are fragmented and
the corresponding MS2 spectra measured. Thus, for some of the ions
(identified as MS1 chromatographic peaks) MS2 spectra are available.
These can facilitate the annotation of the respective MS1
chromatographic peaks (or MS1 features after a correspondence analysis).
Spectra for identified chromatographic peaks can be extracted with the
chromPeakSpectra() method. MS2 spectra with their precursor
m/z and retention time within the rt and m/z range of the
chromatographic peak are returned.
library(Spectra)
dda_spectra <- chromPeakSpectra(dda_data, msLevel = 2L)
dda_spectra## MSn data (Spectra) with 142 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 237.869 1812
## 2 2 241.299 1846
## 3 2 325.763 2439
## 4 2 326.583 2446
## 5 2 327.113 2450
## ... ... ... ...
## 138 2 574.725 5110
## 139 2 575.255 5115
## 140 2 596.584 5272
## 141 2 592.424 5236
## 142 2 596.054 5266
## ... 37 more variables/columns.
##
## file(s):
## 3be760a7234d_7861
## Processing:
## Filter: select retention time [230..610] on MS level(s) [Sun Jul 5 13:04:16 2026]
## Filter: select MS level(s) 2 [Sun Jul 5 13:04:25 2026]
## Merge 1 Spectra into one [Sun Jul 5 13:04:25 2026]By default chromPeakSpectra() returns all spectra
associated with a MS1 chromatographic peak, but parameter
method allows to choose and return only one spectrum per
peak (have a look at the ?chromPeakSpectra help page for
more details). Also, it would be possible to extract MS1 spectra for
each peak by specifying msLevel = 1L in the call above
(e.g. to evaluate the full MS1 signal at the peak’s apex position).
The returned Spectra contains also the reference to the
respective chromatographic peak as additional spectra variable
"chrom_peak_id" that contains the identifier for the
chromatographic peak (i.e. its row name in the chromPeaks
matrix).
dda_spectra$chrom_peak_id## [1] "CP004" "CP004" "CP005" "CP005" "CP006" "CP006" "CP008" "CP008" "CP011"
## [10] "CP011" "CP012" "CP012" "CP013" "CP013" "CP013" "CP013" "CP014" "CP014"
## [19] "CP014" "CP014" "CP018" "CP022" "CP022" "CP022" "CP025" "CP025" "CP025"
## [28] "CP025" "CP026" "CP026" "CP026" "CP026" "CP033" "CP033" "CP034" "CP034"
## [37] "CP034" "CP034" "CP034" "CP035" "CP035" "CP035" "CP041" "CP041" "CP041"
## [46] "CP042" "CP042" "CP042" "CP043" "CP047" "CP047" "CP049" "CP049" "CP049"
## [55] "CP049" "CP050" "CP050" "CP050" "CP051" "CP051" "CP051" "CP054" "CP055"
## [64] "CP055" "CP055" "CP056" "CP056" "CP056" "CP056" "CP056" "CP060" "CP060"
## [73] "CP060" "CP060" "CP064" "CP064" "CP065" "CP065" "CP066" "CP066" "CP069"
## [82] "CP069" "CP069" "CP070" "CP070" "CP070" "CP072" "CP072" "CP072" "CP073"
## [91] "CP074" "CP074" "CP074" "CP074" "CP075" "CP075" "CP075" "CP077" "CP077"
## [100] "CP077" "CP079" "CP079" "CP079" "CP079" "CP080" "CP080" "CP080" "CP081"
## [109] "CP086" "CP086" "CP086" "CP086" "CP086" "CP088" "CP088" "CP088" "CP089"
## [118] "CP089" "CP091" "CP091" "CP093" "CP093" "CP094" "CP094" "CP094" "CP095"
## [127] "CP095" "CP095" "CP096" "CP096" "CP096" "CP098" "CP098" "CP098" "CP098"
## [136] "CP098" "CP099" "CP099" "CP099" "CP100" "CP101" "CP101"Some information about the chromatographic peak can also be added to
the returned Spectra object using the
chrompeakColumns parameter in
chromPeakSpectra(). By default, the m/z and retention time
of the chromatographic peak are added to the spectra metadata.
dda_spectra$chrom_peak_mz## [1] 219.0957 219.0957 153.0659 153.0659 235.1447 235.1447 298.2751 298.2751
## [9] 284.0545 284.0545 306.0364 306.0364 589.0833 589.0833 589.0833 589.0833
## [17] 227.9913 227.9913 227.9913 227.9913 300.0312 278.0916 278.0916 278.0916
## [25] 256.1102 256.1102 256.1102 256.1102 224.0836 224.0836 224.0836 224.0836
## [33] 309.9980 309.9980 376.0385 376.0385 376.0385 376.0385 376.0385 308.0013
## [41] 308.0013 308.0013 286.1806 286.1806 286.1806 292.1215 292.1215 292.1215
## [49] 313.0393 276.0817 276.0817 326.0952 326.0952 326.0952 326.0952 629.2013
## [57] 629.2013 629.2013 607.2193 607.2193 607.2193 291.1192 289.1217 289.1217
## [65] 289.1217 304.1133 304.1133 304.1133 304.1133 304.1133 382.9727 382.9727
## [73] 382.9727 382.9727 249.0247 249.0247 603.1183 603.1183 291.0719 291.0719
## [81] 313.0540 313.0540 313.0540 231.0145 231.0145 231.0145 386.0546 386.0546
## [89] 386.0546 152.0504 194.0969 194.0969 194.0969 194.0969 364.0734 364.0734
## [97] 364.0734 238.1221 238.1221 238.1221 349.1518 349.1518 349.1518 349.1518
## [105] 327.1698 327.1698 327.1698 205.0970 306.1636 306.1636 306.1636 306.1636
## [113] 306.1636 354.0895 354.0895 354.0895 230.9875 230.9875 216.1414 216.1414
## [121] 395.0813 395.0813 284.1641 284.1641 284.1641 331.2081 331.2081 331.2081
## [129] 330.2067 330.2067 330.2067 330.9940 330.9940 330.9940 330.9940 330.9940
## [137] 373.0414 373.0414 373.0414 313.0391 411.1123 411.1123Note also that with return.type = "List" a list parallel
to the chromPeaks matrix would be returned, i.e. each
element in that list would contain the spectra for the chromatographic
peak with the same index. Such data representation might eventually
simplify further processing.
We next use the MS2 information to aid in the annotation of a chromatographic peak. As an example we use a chromatographic peak of an ion with an m/z of 304.1131 which we extract in the code block below.
ex_mz <- 304.1131
chromPeaks(dda_data, mz = ex_mz, ppm = 20)## mz mzmin mzmax rt rtmin rtmax into intb
## CP056 304.1133 304.1126 304.1143 425.024 417.985 441.773 13040.8 13007.79
## maxo sn sample
## CP056 3978.987 232 1A search of potential ions with a similar m/z in a reference database (e.g. Metlin) returned a large list of potential hits, most with a very small ppm. For two of the hits, Flumazenil (Metlin ID 2724) and Fenamiphos (Metlin ID 72445) experimental MS2 spectra are available. Thus, we could match the MS2 spectrum for the identified chromatographic peak against these to annotate our ion. Below we extract all MS2 spectra that were associated with the candidate chromatographic peak using the ID of the peak in the present data set.
ex_id <- rownames(chromPeaks(dda_data, mz = ex_mz, ppm = 20))
ex_spectra <- dda_spectra[dda_spectra$chrom_peak_id == ex_id]
ex_spectra## MSn data (Spectra) with 5 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 418.926 3505
## 2 2 419.306 3510
## 3 2 423.036 3582
## 4 2 423.966 3603
## 5 2 424.296 3609
## ... 37 more variables/columns.
##
## file(s):
## 3be760a7234d_7861
## Processing:
## Filter: select retention time [230..610] on MS level(s) [Sun Jul 5 13:04:16 2026]
## Filter: select MS level(s) 2 [Sun Jul 5 13:04:25 2026]
## Merge 1 Spectra into one [Sun Jul 5 13:04:25 2026]There are 5 MS2 spectra representing fragmentation of the ion(s)
measured in our candidate chromatographic peak. We next reduce this to a
single MS2 spectrum using the combineSpectra() method
employing the combinePeaks() function to determine which
peaks to keep in the resulting spectrum (have a look at the
?combinePeaks help page for details). Parameter
f allows to specify which spectra in the input object
should be combined into one. Note that this combination of multiple
fragment spectra into a single spectrum might not be generally the best
approach or suggested for all types of data.
ex_spectrum <- combineSpectra(ex_spectra, FUN = combinePeaks, ppm = 20,
peaks = "intersect", minProp = 0.8,
intensityFun = median, mzFun = median,
f = ex_spectra$chrom_peak_id)## Warning in FUN(X[[i]], ...): 'combinePeaks' for lists of peak matrices is
## deprecated; please use 'combinePeaksData' instead.
ex_spectrum## MSn data (Spectra) with 1 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 418.926 3505
## ... 37 more variables/columns.
## Processing:
## Filter: select retention time [230..610] on MS level(s) [Sun Jul 5 13:04:16 2026]
## Filter: select MS level(s) 2 [Sun Jul 5 13:04:25 2026]
## Merge 1 Spectra into one [Sun Jul 5 13:04:25 2026]
## ...1 more processings. Use 'processingLog' to list all.Mass peaks from all input spectra with a difference in m/z smaller 20
ppm (parameter ppm) were combined into one peak and the
median m/z and intensity is reported for these. Due to parameter
minProp = 0.8, the resulting MS2 spectrum contains only
peaks that were present in 80% of the input spectra.
A plot of this consensus spectrum is shown below.
plotSpectra(ex_spectrum)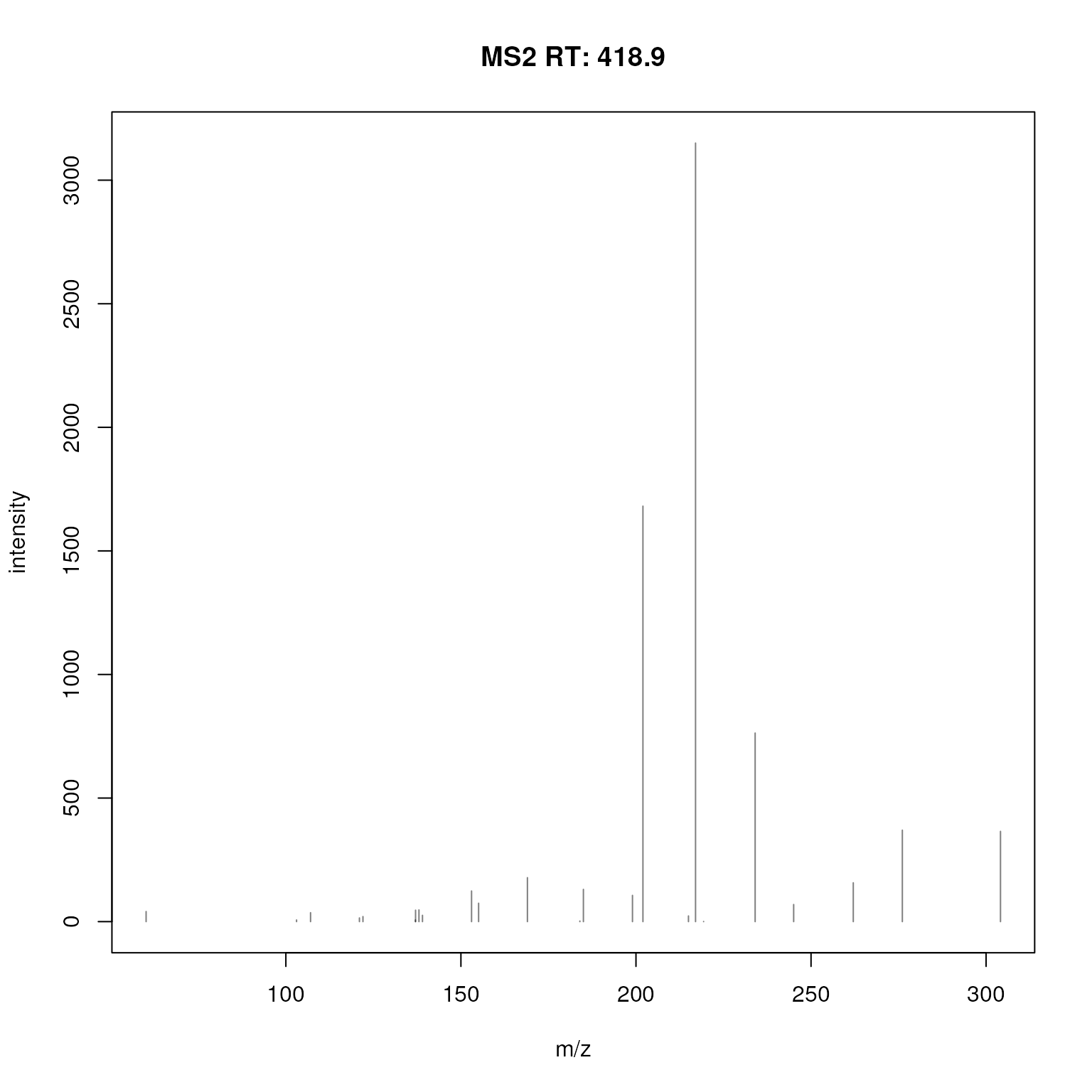
Consensus MS2 spectrum created from all measured MS2 spectra for ions of chromatographic peak CP53.
We could now match the consensus spectrum against a database of MS2 spectra. In our example we simply load MS2 spectra for the two compounds with matching m/z exported from Metlin. For each of the compounds MS2 spectra created with collision energies of 0V, 10V, 20V and 40V are available. Below we import the respective data and plot our candidate spectrum against the MS2 spectra of Flumanezil and Fenamiphos (from a collision energy of 20V). To import files in MGF format we have to load the MsBackendMgf Bioconductor package which adds MGF file support to the Spectra package.
Prior plotting we scale our experimental spectra to replace all peak intensities with values relative to the maximum peak intensity (which is set to a value of 100).
scale_fun <- function(z, ...) {
z[, "intensity"] <- z[, "intensity"] /
max(z[, "intensity"], na.rm = TRUE) * 100
z
}
ex_spectrum <- addProcessing(ex_spectrum, FUN = scale_fun)
library(MsBackendMgf)
flumanezil <- Spectra(
system.file("mgf", "metlin-2724.mgf", package = "xcms"),
source = MsBackendMgf())## Start data import from 1 files ... done
fenamiphos <- Spectra(
system.file("mgf", "metlin-72445.mgf", package = "xcms"),
source = MsBackendMgf())## Start data import from 1 files ... done
par(mfrow = c(1, 2))
plotSpectraMirror(ex_spectrum, flumanezil[3], main = "against Flumanezil",
ppm = 40)
plotSpectraMirror(ex_spectrum, fenamiphos[3], main = "against Fenamiphos",
ppm = 40)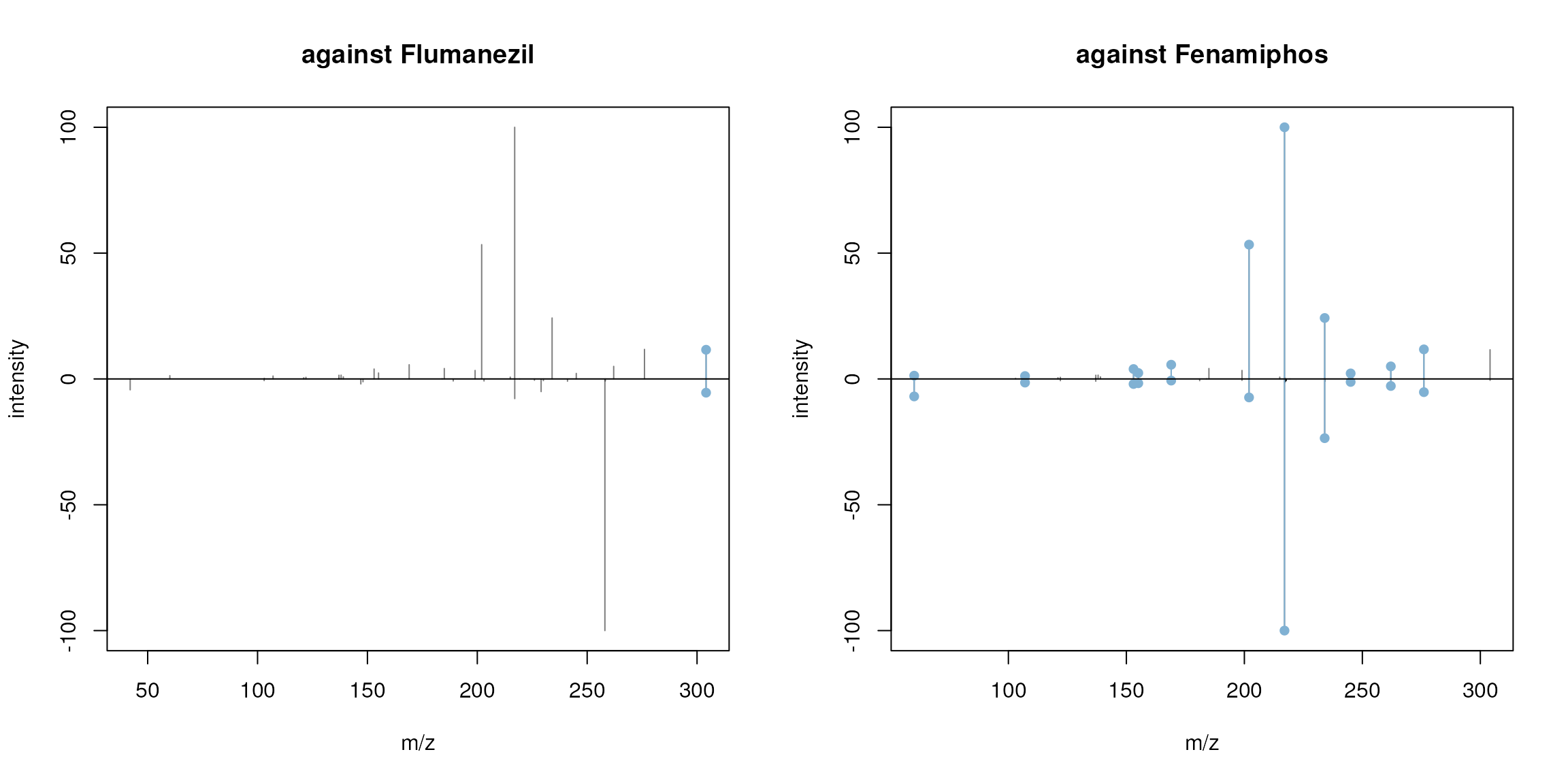
Mirror plots for the candidate MS2 spectrum against Flumanezil (left) and Fenamiphos (right). The upper panel represents the candidate MS2 spectrum, the lower the target MS2 spectrum. Matching peaks are indicated with a dot.
Our candidate spectrum matches Fenamiphos, thus, our example
chromatographic peak represents signal measured for this compound. In
addition to plotting the spectra, we can also calculate similarities
between them with the compareSpectra() method (which uses
by default the normalized dot-product to calculate the similarity).
compareSpectra(ex_spectrum, flumanezil, ppm = 40)## [1] 4.520957e-02 3.283806e-02 2.049379e-03 3.374354e-05
compareSpectra(ex_spectrum, fenamiphos, ppm = 40)## [1] 0.1326234432 0.4879399946 0.7198406271 0.3997922658 0.0004876129
## [6] 0.0028408885 0.0071030051 0.0053809736Clearly, the candidate spectrum does not match Flumanezil, while it
has a high similarity to Fenamiphos. While we performed here the
MS2-based annotation on a single chromatographic peak, this could be
easily extended to the full list of MS2 spectra (returned by
chromPeakSpectra()) for all chromatographic peaks in an
experiment. See also here or here for
alternative tutorials on matching experimental fragment spectra against
a reference.
In the present example we used only a single data file and we did
thus not need to perform a sample alignment and correspondence analysis.
These tasks could however be performed similarly to plain LC-MS
data, retention times of recorded MS2 spectra would however also be
adjusted during alignment based on the MS1 data. After correspondence
analysis (peak grouping) MS2 spectra for features can be
extracted with the featureSpectra() function which returns
all MS2 spectra associated with any chromatographic peak of a
feature.
Note also that this workflow can be included into the Feature-Based Molecular Networking FBMN to match MS2 spectra against GNPS. See here for more details and examples.
DIA (SWATH) data analysis
In this section we analyze a small SWATH data set consisting of a
single mzML file with data from the same sample analyzed in the previous
section but recorded in SWATH mode. We again read the data with the
readMsExperiment() function. The resulting object will
contain all recorded MS1 and MS2 spectra in the specified file. Similar
to the previous data file, we filter the file to signal between 230 and
610 seconds.
swath_file <- PestMix1_SWATH.mzML()
swath_data <- readMsExperiment(swath_file)
swath_data <- filterRt(swath_data, rt = c(230, 610))Below we determine the number of MS level 1 and 2 spectra in the present data set.
##
## 1 2
## 422 3378As described in the introduction, in SWATH mode all ions within
pre-defined isolation windows are fragmented and MS2 spectra measured.
The definition of these isolation windows (SWATH pockets) is imported
from the mzML files and available as additional spectra
variables. Below we inspect the respective information for the
first few spectra. The upper and lower isolation window m/z is available
with spectra variables "isolationWindowLowerMz" and
"isolationWindowUpperMz" respectively and the
target m/z of the isolation window with
"isolationWindowTargetMz". We can use the
spectraData() function to extract this information from the
spectra within our swath_data object.
spectra(swath_data) |>
spectraData(c("isolationWindowTargetMz", "isolationWindowLowerMz",
"isolationWindowUpperMz", "msLevel", "rtime")) |>
head()## DataFrame with 6 rows and 5 columns
## isolationWindowTargetMz isolationWindowLowerMz isolationWindowUpperMz
## <numeric> <numeric> <numeric>
## 1 299.10 283.5 314.7
## 2 329.80 313.7 345.9
## 3 367.35 344.9 389.8
## 4 601.85 388.8 814.9
## 5 NA NA NA
## 6 163.75 139.5 188.0
## msLevel rtime
## <integer> <numeric>
## 1 2 230.073
## 2 2 230.170
## 3 2 230.267
## 4 2 230.364
## 5 1 230.459
## 6 2 230.585We could also access these variables directly with the dedicated
isolationWindowLowerMz() and
isolationWindowUpperMz() functions.
head(isolationWindowLowerMz(spectra(swath_data)))## [1] 283.5 313.7 344.9 388.8 NA 139.5
head(isolationWindowUpperMz(spectra(swath_data)))## [1] 314.7 345.9 389.8 814.9 NA 188.0In the present data set we use the value of the isolation window target m/z to define the individual SWATH pockets. Below we list the number of spectra that are recorded in each pocket/isolation window.
table(isolationWindowTargetMz(spectra(swath_data)))##
## 163.75 208.95 244.05 270.85 299.1 329.8 367.35 601.85
## 422 422 422 422 423 423 422 422We have thus between 422 and 423 MS2 spectra measured in each isolation window.
To inspect the data we can also extract chromatograms from both the
measured MS1 as well as MS2 data. For MS2 data we have to set parameter
msLevel = 2L and, for SWATH data, in addition also specify
the isolation window from which we want to extract the data. Below we
extract the TIC of the MS1 data and of one of the isolation windows
(isolation window target m/z of 270.85) and plot these.
tic_ms1 <- chromatogram(swath_data, msLevel = 1L, aggregationFun = "sum")
tic_ms2 <- chromatogram(swath_data, msLevel = 2L, aggregationFun = "sum",
isolationWindowTargetMz = 270.85)
par(mfrow = c(2, 1))
plot(tic_ms1, main = "MS1")
plot(tic_ms2, main = "MS2, isolation window m/z 270.85")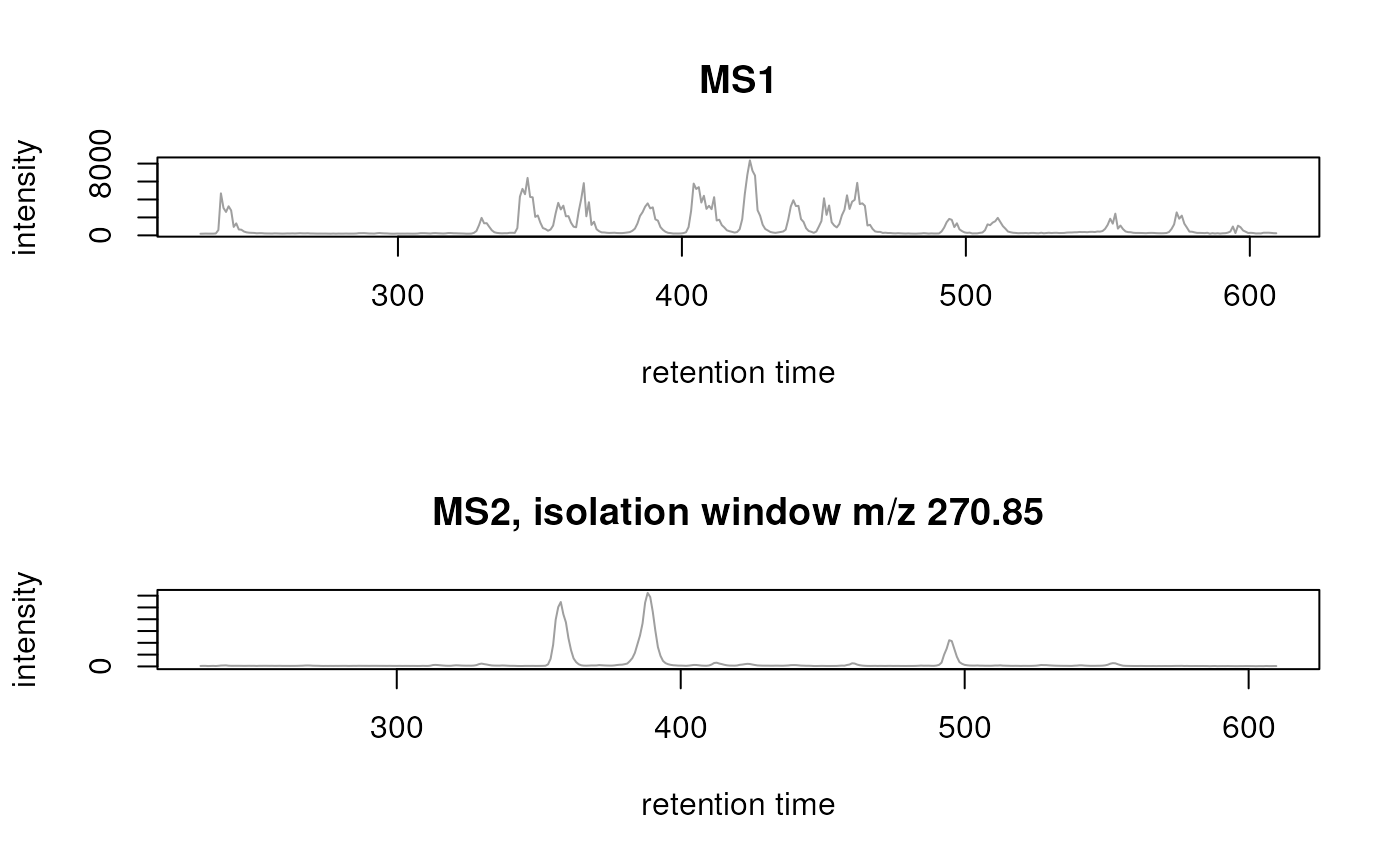
TIC for MS1 (upper panel) and MS2 data from the isolation window with target m/z 270.85 (lower panel).
Without specifying the isolationWindowTargetMz
parameter, all MS2 spectra would be considered in the chromatogram
extraction which would result in a chimeric chromatogram such
as the one shown below:
tic_all_ms2 <- chromatogram(swath_data, msLevel = 2L, aggregationFun = "sum")
plot(tic_all_ms2, main = "MS2, all isolation windows")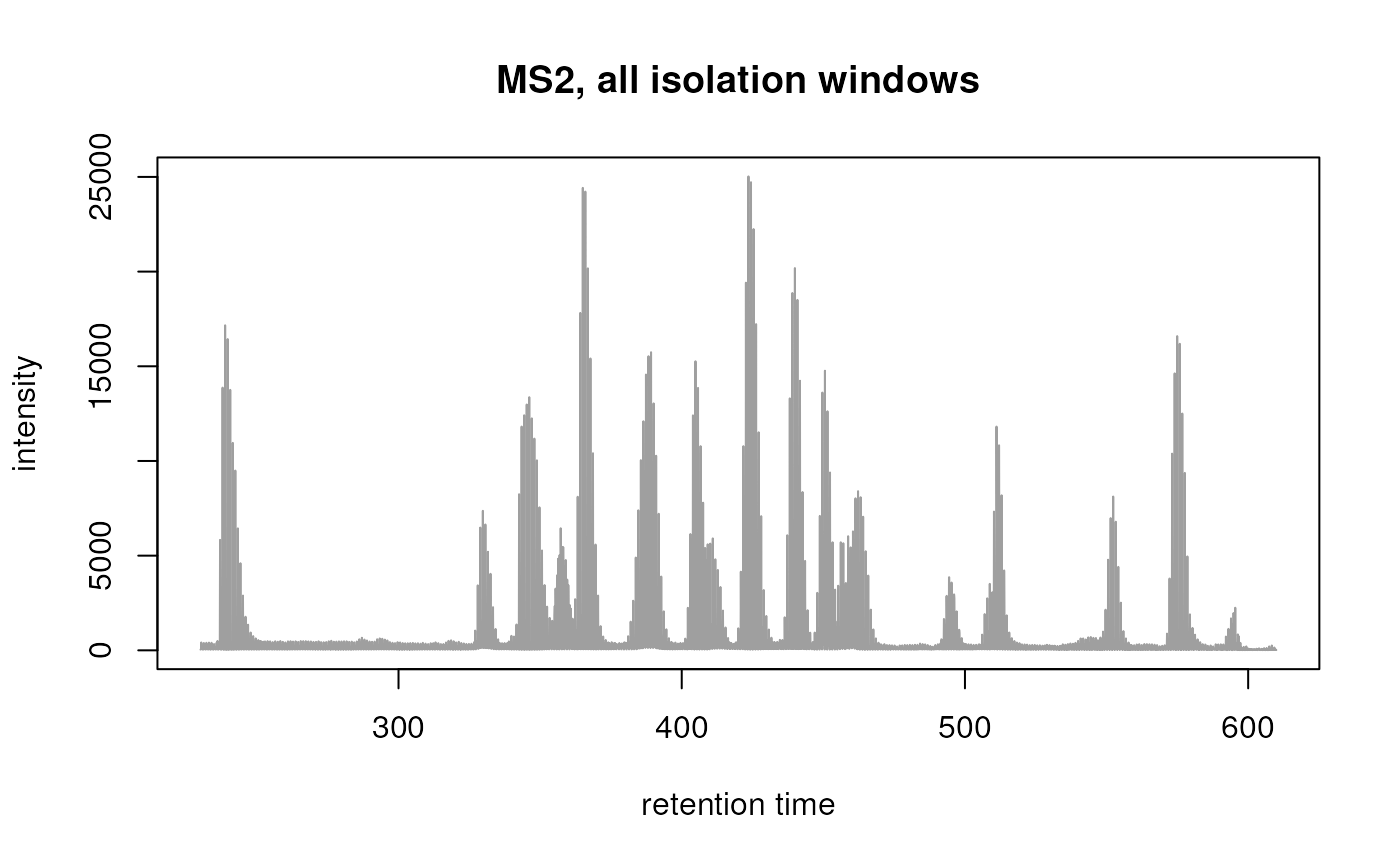
TIC considering all MS2 spectra (from all isolation windows).
For MS2 data without specific, different, m/z
isolation windows (such as e.g. Waters MSe data) parameter
isolationWindowTargetMz can be omitted in the
chromatograms() call in which case, as already stated
above, all MS2 spectra are considered in the chromatogram calculation.
Alternatively, if the isolation window is not provided or specified in
the original data files, it would be possible to manually define a value
for this spectra variable, such as in the example below (from which the
code is however not evaluated) were we assign the value of the precursor
m/z to the spectra’s isolation window target m/z.
spectra(swath_data)$isolationWindowTargetMz <- precursorMz(spectra(swath_data))Chromatographic peak detection in MS1 and MS2 data
Similar to a conventional LC-MS analysis, we perform first a
chromatographic peak detection (on the MS level 1 data) with the
findChromPeaks() method. Below we define the settings for a
centWave-based peak detection and perform the analysis.
cwp <- CentWaveParam(snthresh = 5, noise = 100, ppm = 10,
peakwidth = c(3, 30))
swath_data <- findChromPeaks(swath_data, param = cwp)
swath_data## Object of class XcmsExperiment
## Spectra: MS1 (422) MS2 (3378)
## Experiment data: 1 sample(s)
## Sample data links:
## - spectra: 1 sample(s) to 3800 element(s).
## xcms results:
## - chromatographic peaks: 62 in MS level(s): 1Next we perform a chromatographic peak detection in MS level 2 data
separately for each individual isolation window. We use the
findChromPeaksIsolationWindow() function employing the same
peak detection algorithm reducing however the required signal-to-noise
ratio. The isolationWindow parameter allows to specify
which MS2 spectra belong to which isolation window and hence defines in
which set of MS2 spectra chromatographic peak detection should be
performed. As a default the "isolationWindowTargetMz"
variable of the object’s spectra is used.
cwp <- CentWaveParam(snthresh = 3, noise = 10, ppm = 10,
peakwidth = c(3, 30))
swath_data <- findChromPeaksIsolationWindow(swath_data, param = cwp)## Warning in rbindlistWithRownames(lapply(res[lns > 0], function(z) {: Dropping
## rownames: duplicated rownames present or rownames not available for all
## data.frames
swath_data## Object of class XcmsExperiment
## Spectra: MS1 (422) MS2 (3378)
## Experiment data: 1 sample(s)
## Sample data links:
## - spectra: 1 sample(s) to 3800 element(s).
## xcms results:
## - chromatographic peaks: 370 in MS level(s): 1, 2The findChromPeaksIsolationWindow() function added all
peaks identified in the individual isolation windows to the
chromPeaks matrix containing already the MS1
chromatographic peaks. These newly added peaks can be identified through
the "isolationWindow" column in the object’s
chromPeakData.
chromPeakData(swath_data)## DataFrame with 370 rows and 6 columns
## ms_level is_filled isolationWindow isolationWindowTargetMZ
## <integer> <logical> <numeric> <numeric>
## CP01 1 FALSE NA NA
## CP02 1 FALSE NA NA
## CP03 1 FALSE NA NA
## CP04 1 FALSE NA NA
## CP05 1 FALSE NA NA
## ... ... ... ... ...
## CP366 2 FALSE 601.85 601.85
## CP367 2 FALSE 601.85 601.85
## CP368 2 FALSE 601.85 601.85
## CP369 2 FALSE 601.85 601.85
## CP370 2 FALSE 601.85 601.85
## isolationWindowLowerMz isolationWindowUpperMz
## <numeric> <numeric>
## CP01 NA NA
## CP02 NA NA
## CP03 NA NA
## CP04 NA NA
## CP05 NA NA
## ... ... ...
## CP366 388.8 814.9
## CP367 388.8 814.9
## CP368 388.8 814.9
## CP369 388.8 814.9
## CP370 388.8 814.9Below we count the number of chromatographic peaks identified within each isolation window (the number of chromatographic peaks identified in MS1 is 62).
table(chromPeakData(swath_data)$isolationWindow)##
## 163.75 208.95 244.05 270.85 299.1 329.8 367.35 601.85
## 2 38 32 14 105 23 62 32We thus successfully identified chromatographic peaks in the different MS levels and isolation windows. As a next step we have to identify which of the measured signals represents data from the same original compound to reconstruct fragment spectra for each MS1 signal (chromatographic peak).
Reconstruction of MS2 spectra
Identifying the signal of the fragment ions for the precursor measured by each MS1 chromatographic peak is a non-trivial task. The MS2 spectrum of the fragment ion for each MS1 chromatographic peak has to be reconstructed from the available MS2 signal (i.e. the chromatographic peaks identified in MS level 2). For SWATH data, fragment ion signal should be present in the same isolation window that contains the m/z of the precursor ion and the chromatographic peak shape of the MS2 chromatographic peaks of fragment ions of a specific precursor should have a similar retention time and peak shape than the precursor’s MS1 chromatographic peak.
After detection of MS1 and MS2 chromatographic peaks has been
performed, we can reconstruct the MS2 spectra using the
reconstructChromPeakSpectra() function. This function
defines an MS2 spectrum for each MS1 chromatographic peak based on the
following approach:
- Identify MS2 chromatographic peaks in the isolation window
containing the m/z of the ion (the MS1 chromatographic peak) that have
approximately the same retention time than the MS1 chromatographic peak
(the accepted difference in retention time can be defined with the
diffRtparameter). - Extract the MS1 chromatographic peak and all MS2 chromatographic
peaks identified by the previous step and correlate the peak shapes of
the candidate MS2 chromatographic peaks with the shape of the MS1 peak.
MS2 chromatographic peaks with a correlation coefficient larger than
minCorare retained. - Reconstruct the MS2 spectrum using the m/z of all above selected MS2 chromatographic peaks and their intensity; each MS2 chromatographic peak selected for an MS1 peak will thus represent one mass peak in the reconstructed spectrum.
To illustrate this process we perform the individual steps on the example of fenamiphos (exact mass 303.105800777 and m/z of [M+H]+ adduct 304.113077). As a first step we extract the chromatographic peak for this ion.
fenamiphos_mz <- 304.113077
fenamiphos_ms1_peak <- chromPeaks(swath_data, mz = fenamiphos_mz, ppm = 2)
fenamiphos_ms1_peak## mz mzmin mzmax rt rtmin rtmax into intb
## CP34 304.1124 304.1121 304.1126 423.945 419.445 428.444 10697.34 10688.34
## maxo sn sample
## CP34 2401.849 618 1Next we identify all MS2 chromatographic peaks that were identified
in the isolation window containing the m/z of fenamiphos. The
information on the isolation window in which a chromatographic peak was
identified is available in the chromPeakData.
keep <- chromPeakData(swath_data)$isolationWindowLowerMz < fenamiphos_mz &
chromPeakData(swath_data)$isolationWindowUpperMz > fenamiphos_mzWe also require the retention time of the MS2 chromatographic peaks to be similar to the retention time of the MS1 peak and extract the corresponding peak information. We thus below select all chromatographic peaks for which the retention time range contains the retention time of the apex position of the MS1 chromatographic peak.
keep <- keep &
chromPeaks(swath_data)[, "rtmin"] < fenamiphos_ms1_peak[, "rt"] &
chromPeaks(swath_data)[, "rtmax"] > fenamiphos_ms1_peak[, "rt"]
fenamiphos_ms2_peak <- chromPeaks(swath_data)[which(keep), ]In total 24 MS2 chromatographic peaks match all the above conditions.
Next we extract the ion chromatogram of the MS1 peak and of all selected
candidate MS2 signals. To ensure chromatograms are extracted from
spectra in the correct isolation window we need to specify the
respective isolation window by passing its isolation window target m/z
to the chromatogram() function (in addition to setting
msLevel = 2). This can be done by either getting the
isolationWindowTargetMz of the spectra after the data was
subset using filterIsolationWindow() (as done below) or by
selecting the isolationWindowTargetMz closest to the m/z of
the compound of interest.
rtr <- fenamiphos_ms1_peak[, c("rtmin", "rtmax")]
mzr <- fenamiphos_ms1_peak[, c("mzmin", "mzmax")]
fenamiphos_ms1_chr <- chromatogram(swath_data, rt = rtr, mz = mzr)## Extracting chromatographic data## Processing chromatographic peaks
rtr <- fenamiphos_ms2_peak[, c("rtmin", "rtmax")]
mzr <- fenamiphos_ms2_peak[, c("mzmin", "mzmax")]
## Get the isolationWindowTargetMz for spectra containing the m/z of the
## compound of interest
swath_data |>
filterIsolationWindow(mz = fenamiphos_mz) |>
spectra() |>
isolationWindowTargetMz() |>
table()##
## 299.1
## 423The target m/z of the isolation window containing the m/z of interest
is thus 299.1 and we can use this in the chromatogram()
call below to extract the data from the correct (MS2) spectra.
fenamiphos_ms2_chr <- chromatogram(
swath_data, rt = rtr, mz = mzr, msLevel = 2L,
isolationWindowTargetMz = rep(299.1, nrow(rtr)))## Extracting chromatographic data## Processing chromatographic peaksWe can now plot the extracted ion chromatogram of the MS1 and the extracted MS2 data.
plot(rtime(fenamiphos_ms1_chr[1, 1]),
intensity(fenamiphos_ms1_chr[1, 1]),
xlab = "retention time [s]", ylab = "intensity", pch = 16,
ylim = c(0, 5000), col = "blue", type = "b", lwd = 2)
#' Add data from all MS2 peaks
tmp <- lapply(fenamiphos_ms2_chr@.Data,
function(z) points(rtime(z), intensity(z),
col = "#00000080",
type = "b", pch = 16))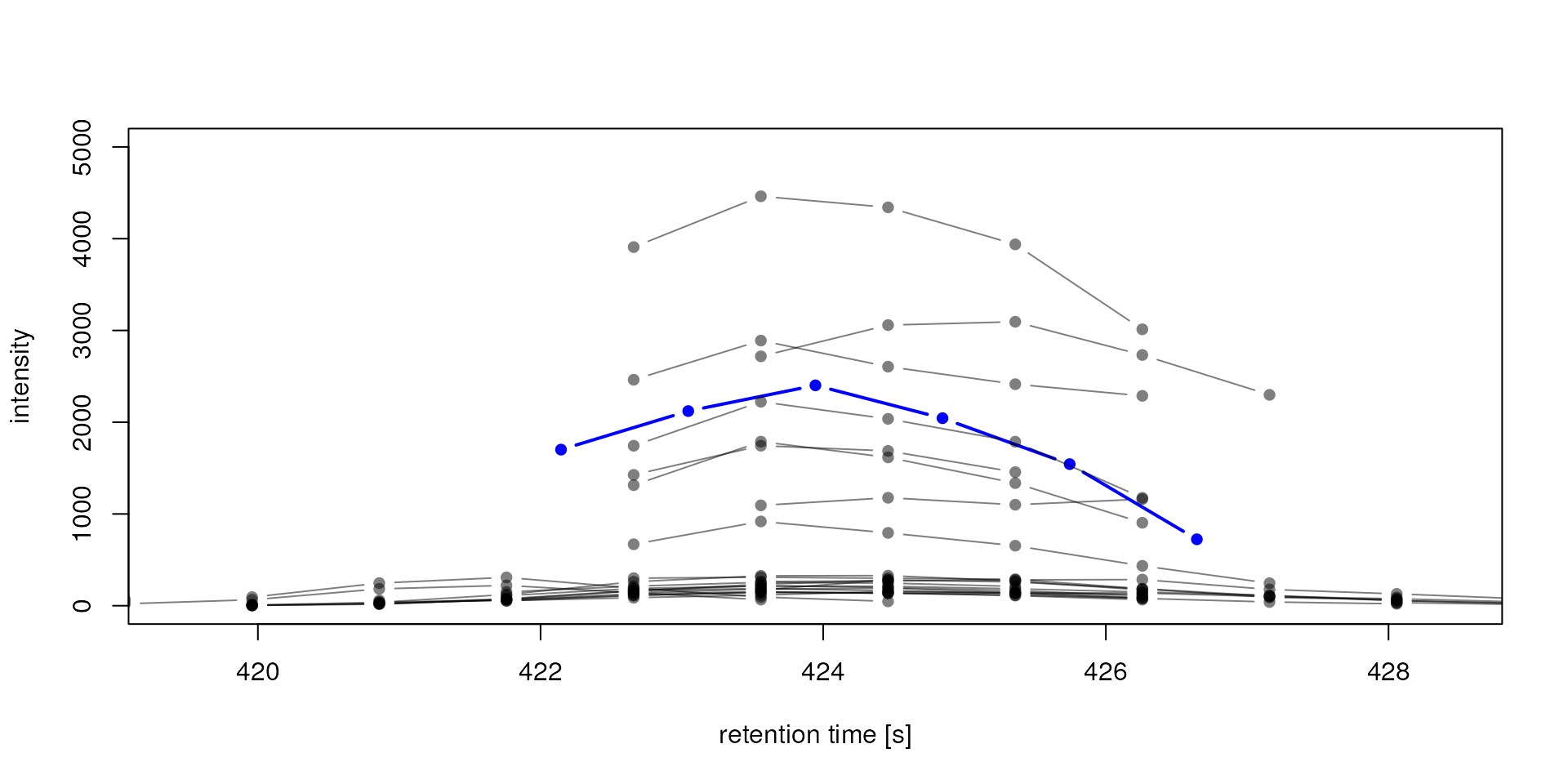
Extracted ion chromatograms for Fenamiphos from MS1 (blue) and potentially related signal in MS2 (grey).
Next we can calculate correlations between the peak shapes of each
MS2 chromatogram with the MS1 peak. We illustrate this process on the
example of one MS2 chromatographic peaks. Note that, because MS1 and MS2
spectra are recorded consecutively, the retention times of the
individual data points will differ between the MS2 and MS1
chromatographic data and data points have thus to be matched (aligned)
before performing the correlation analysis. This is done automatically
by the correlate() function. See the help for the
align method for more information on alignment options.
compareChromatograms(fenamiphos_ms2_chr[1, 1],
fenamiphos_ms1_chr[1, 1],
ALIGNFUNARGS = list(method = "approx"))## [1] 0.9997871After identifying the MS2 chromatographic peaks with shapes of enough
high similarity to the MS1 chromatographic peaks, an MS2 spectrum could
be reconstructed based on the m/z and intensities of the MS2
chromatographic peaks (i.e., using their "mz" and
"maxo" or "into" values).
Instead of performing this assignment of MS2 signal to MS1
chromatographic peaks manually as above, we can use the
reconstructChromPeakSpectra() function that performs the
exact same steps for all MS1 chromatographic peaks in a DIA data set.
Below we use this function to reconstruct MS2 spectra for our example
data requiring a peak shape correlation higher than 0.9
between the candidate MS2 chromatographic peak and the target MS1
chromatographic peak.
swath_spectra <- reconstructChromPeakSpectra(swath_data, minCor = 0.9)
swath_spectra## MSn data (Spectra) with 62 spectra in a MsBackendMemory backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## CP01 2 239.458 NA
## CP02 2 240.358 NA
## CP03 2 329.577 NA
## CP04 2 329.771 NA
## CP05 2 346.164 NA
## ... ... ... ...
## CP58 2 551.735 NA
## CP59 2 551.735 NA
## CP60 2 575.134 NA
## CP61 2 575.134 NA
## CP62 2 574.942 NA
## ... 20 more variables/columns.
## Processing:
## Merge 1 Spectra into one [Sun Jul 5 13:04:33 2026]As a result we got a Spectra object of length equal to
the number of MS1 peaks in our data. The length of a spectrum represents
the number of peaks it contains. Thus, a length of 0 indicates that no
matching peak (MS2 signal) could be found for the respective MS1
chromatographic peak.
lengths(swath_spectra)## [1] 0 0 1 1 1 0 0 0 0 0 0 0 3 0 3 4 0 3 0 1 0 9 14 1 0
## [26] 0 15 4 1 1 2 4 6 15 12 11 2 4 13 0 0 0 0 1 2 0 1 0 0 0
## [51] 3 0 2 1 7 7 0 0 0 0 0 2For reconstructed spectra additional annotations are available such
as the IDs of the MS2 chromatographic peaks from which the spectrum was
reconstructed ("ms2_peak_id") as well as the correlation
coefficient of their chromatographic peak shape with the precursor’s
shape ("ms2_peak_cor"). Metadata column
"peak_id" contains the ID of the MS1 chromatographic
peak:
spectraData(swath_spectra, c("peak_id", "ms2_peak_id", "ms2_peak_cor"))## DataFrame with 62 rows and 3 columns
## peak_id ms2_peak_id ms2_peak_cor
## <character> <list> <list>
## CP01 CP01
## CP02 CP02
## CP03 CP03 CP063 0.950582
## CP04 CP04 CP105 0.95157
## CP05 CP05 CP153 0.924545
## ... ... ... ...
## CP58 CP58
## CP59 CP59
## CP60 CP60
## CP61 CP61
## CP62 CP62 CP334,CP329 0.918915,0.911944We next extract the MS2 spectrum for our example peak most likely representing [M+H]+ ions of Fenamiphos using its chromatographic peak ID:
fenamiphos_swath_spectrum <- swath_spectra[
swath_spectra$peak_id == rownames(fenamiphos_ms1_peak)]We can now compare the reconstructed spectrum to the example
consensus spectrum from the DDA experiment in the previous section
(variable ex_spectrum) as well as to the MS2 spectrum for
Fenamiphos from Metlin (with a collision energy of 10V). For better
visualization we normalize also the peak intensities of the
reconstructed SWATH spectrum with the same function we used for the
experimental DDA spectrum.
fenamiphos_swath_spectrum <- addProcessing(fenamiphos_swath_spectrum,
scale_fun)
par(mfrow = c(1, 2))
plotSpectraMirror(fenamiphos_swath_spectrum, ex_spectrum,
ppm = 50, main = "against DDA")
plotSpectraMirror(fenamiphos_swath_spectrum, fenamiphos[2],
ppm = 50, main = "against Metlin")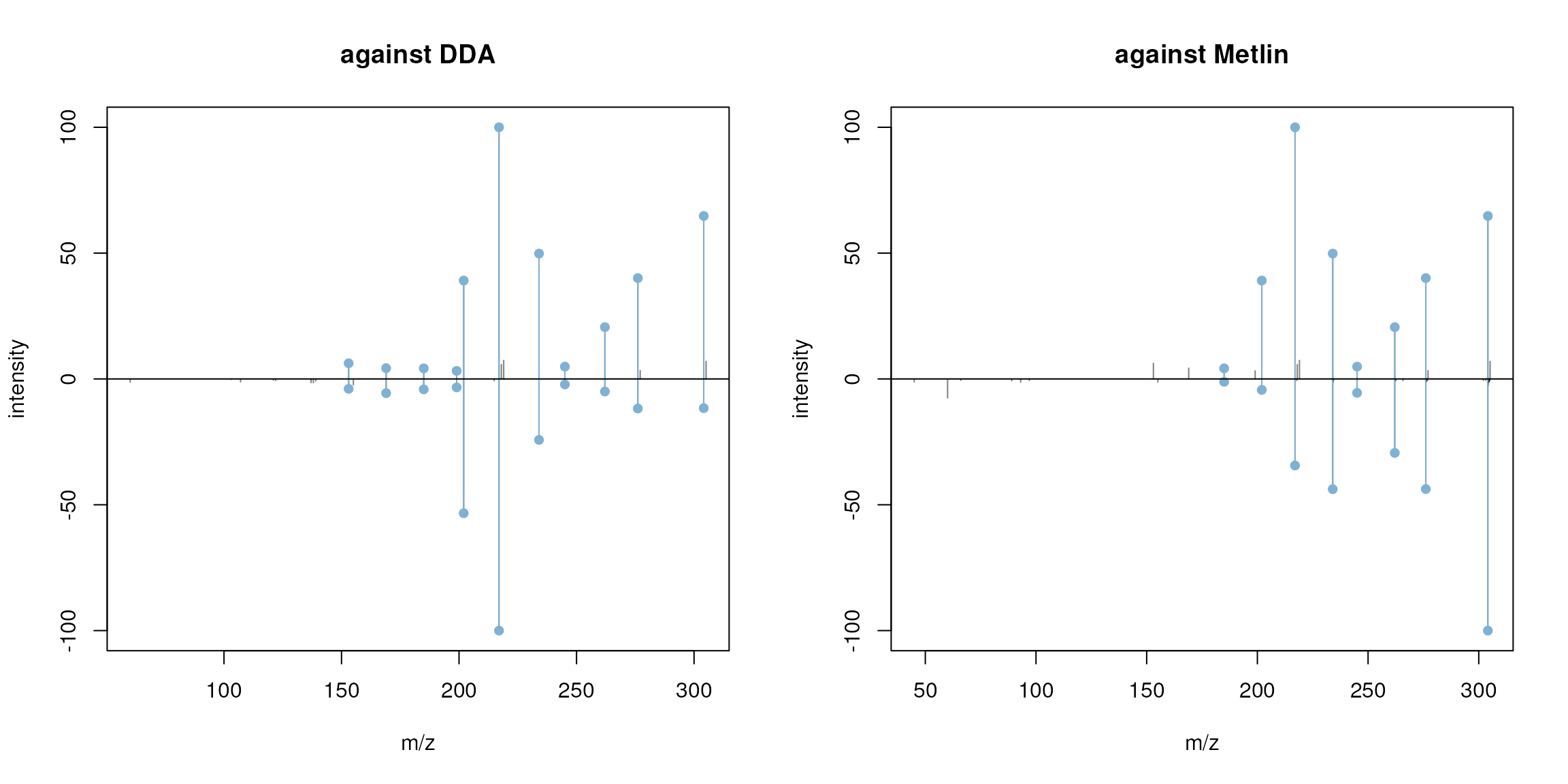
Mirror plot comparing the reconstructed MS2 spectrum for Fenamiphos (upper panel) against the measured spectrum from the DDA data and the Fenamiphhos spectrum from Metlin.
If we wanted to get the EICs for the MS2 chromatographic peaks used
to generate this MS2 spectrum we can use the IDs of these peaks which
are provided with $ms2_peak_id of the result spectrum.
pk_ids <- fenamiphos_swath_spectrum$ms2_peak_id[[1]]
pk_ids## [1] "CP199" "CP201" "CP211" "CP208" "CP200" "CP202" "CP217" "CP215" "CP205"
## [10] "CP212" "CP221" "CP223" "CP213" "CP207" "CP220"With these peak IDs available we can extract their retention time
window and m/z ranges from the chromPeaks matrix and use
the chromatogram() function to extract their EIC. Note
however that for SWATH data we have MS2 signal from different isolation
windows. Thus we have to first filter the swath_data object
by the isolation window containing the precursor m/z with the
filterIsolationWindow() to subset the data to MS2 spectra
related to the ion of interest. In addition, we have to use
msLevel = 2L in the chromatogram() call
because chromatogram() extracts by default only data from
MS1 spectra and we need to specify the target m/z of the isolation
window containing the fragment data from the compound of interest.
rt_range <- chromPeaks(swath_data)[pk_ids, c("rtmin", "rtmax")]
mz_range <- chromPeaks(swath_data)[pk_ids, c("mzmin", "mzmax")]
pmz <- precursorMz(fenamiphos_swath_spectrum)[1]
## Determine the isolation window target m/z
tmz <- swath_data |>
filterIsolationWindow(mz = pmz) |>
spectra() |>
isolationWindowTargetMz() |>
unique()
ms2_eics <- chromatogram(
swath_data, rt = rt_range, mz = mz_range, msLevel = 2L,
isolationWindowTargetMz = rep(tmz, nrow(rt_range)))## Extracting chromatographic data## Processing chromatographic peaksEach row of this ms2_eics contains now the EIC of one of
the MS2 chromatographic peaks. We can also plot these in an overlay
plot.
plotChromatogramsOverlay(ms2_eics)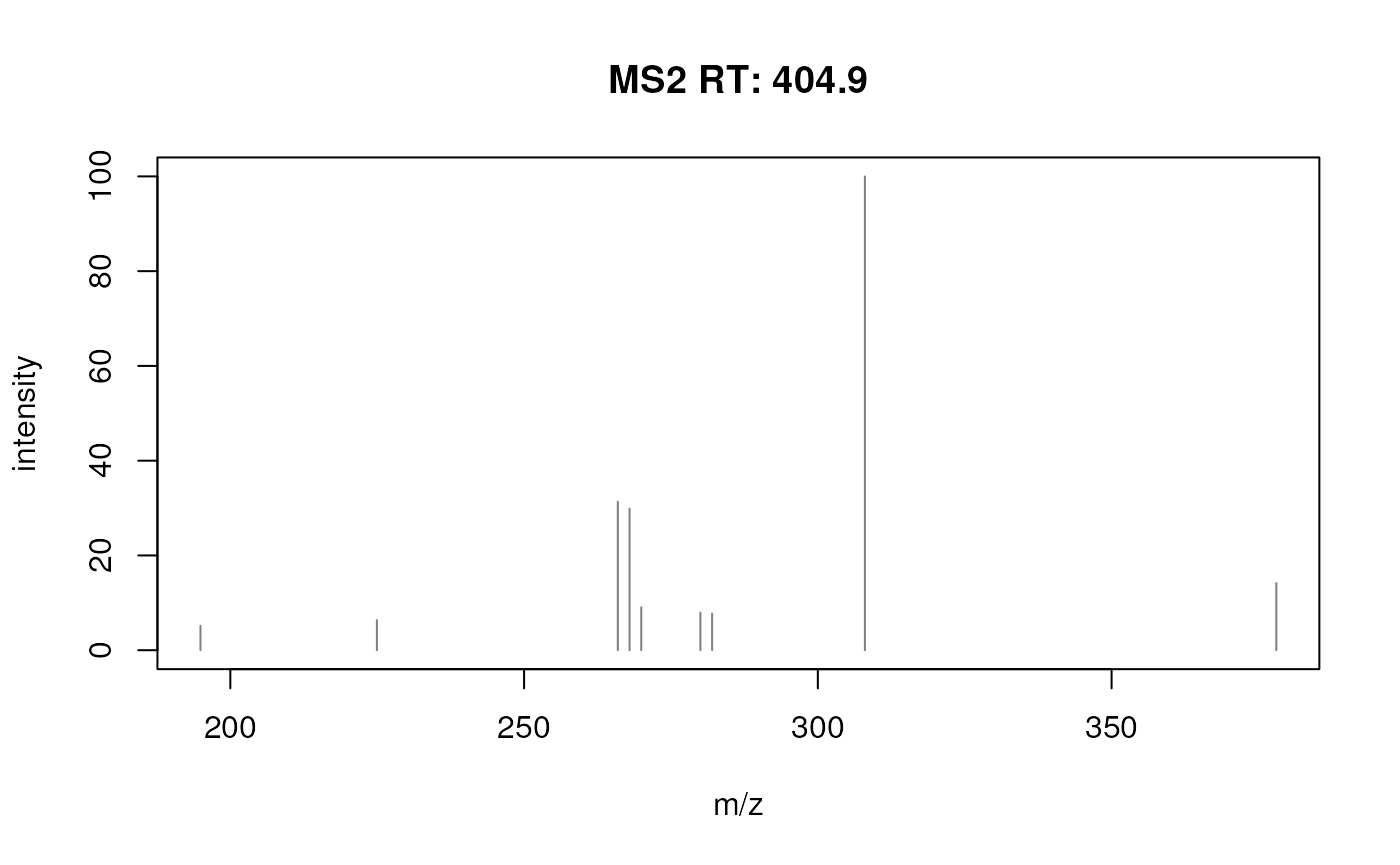
Overlay of EICs of chromatographic peaks used to reconstruct the MS2 spectrum for fenamiphos.
As a second example we analyze the signal from an [M+H]+ ion with an m/z of 376.0381 (which would match Prochloraz). We first identify the MS1 chromatographic peak for that m/z and retrieve the reconstructed MS2 spectrum for that peak.
prochloraz_mz <- 376.0381
prochloraz_ms1_peak <- chromPeaks(swath_data, msLevel = 1L,
mz = prochloraz_mz, ppm = 5)
prochloraz_ms1_peak## mz mzmin mzmax rt rtmin rtmax into intb
## CP22 376.0373 376.037 376.0374 405.046 401.446 409.546 3664.051 3655.951
## maxo sn sample
## CP22 897.3923 278 1
prochloraz_swath_spectrum <- swath_spectra[
swath_spectra$peak_id == rownames(prochloraz_ms1_peak)]
lengths(prochloraz_swath_spectrum)## [1] 9The MS2 spectrum for the (tentative) MS1 signal for prochloraz reconstructed from the SWATH MS2 data has thus 9 peaks.
In addition we identify the corresponding MS1 peak in the DDA data set, extract all measured MS2 chromatographic peaks and build the consensus spectrum from these.
prochloraz_dda_peak <- chromPeaks(dda_data, msLevel = 1L,
mz = prochloraz_mz, ppm = 5)
prochloraz_dda_peak## mz mzmin mzmax rt rtmin rtmax into intb
## CP034 376.0385 376.0378 376.0391 405.295 401.166 410.145 5082.157 5072.77
## maxo sn sample
## CP034 1350.633 310 1The retention times for the chromatographic peaks from the DDA and SWATH data match almost perfectly. Next we get the MS2 spectra for this peak.
prochloraz_dda_spectra <- dda_spectra[
dda_spectra$chrom_peak_id == rownames(prochloraz_dda_peak)]
prochloraz_dda_spectra## MSn data (Spectra) with 5 spectra in a MsBackendMzR backend:
## msLevel rtime scanIndex
## <integer> <numeric> <integer>
## 1 2 401.438 3253
## 2 2 402.198 3259
## 3 2 404.677 3306
## 4 2 405.127 3316
## 5 2 405.877 3325
## ... 37 more variables/columns.
##
## file(s):
## 3be760a7234d_7861
## Processing:
## Filter: select retention time [230..610] on MS level(s) [Sun Jul 5 13:04:16 2026]
## Filter: select MS level(s) 2 [Sun Jul 5 13:04:25 2026]
## Merge 1 Spectra into one [Sun Jul 5 13:04:25 2026]In total 5 spectra were measured, some with a relatively high number of peaks. Next we combine them into a consensus spectrum.
prochloraz_dda_spectrum <- combineSpectra(
prochloraz_dda_spectra, FUN = combinePeaks, ppm = 20,
peaks = "intersect", minProp = 0.8, intensityFun = median, mzFun = median,
f = prochloraz_dda_spectra$chrom_peak_id)## Backend of the input object is read-only, will change that to an 'MsBackendMemory'## Warning in FUN(X[[i]], ...): 'combinePeaks' for lists of peak matrices is
## deprecated; please use 'combinePeaksData' instead.At last we load also the Prochloraz MS2 spectra (for different collision energies) from Metlin.
prochloraz <- Spectra(
system.file("mgf", "metlin-68898.mgf", package = "xcms"),
source = MsBackendMgf())## Start data import from 1 files ... doneTo validate the reconstructed spectrum we plot it against the corresponding DDA spectrum and the MS2 spectrum for Prochloraz (for a collision energy of 10V) from Metlin.
prochloraz_swath_spectrum <- addProcessing(prochloraz_swath_spectrum, scale_fun)
prochloraz_dda_spectrum <- addProcessing(prochloraz_dda_spectrum, scale_fun)
par(mfrow = c(1, 2))
plotSpectraMirror(prochloraz_swath_spectrum, prochloraz_dda_spectrum,
ppm = 40, main = "against DDA")
plotSpectraMirror(prochloraz_swath_spectrum, prochloraz[2],
ppm = 40, main = "against Metlin")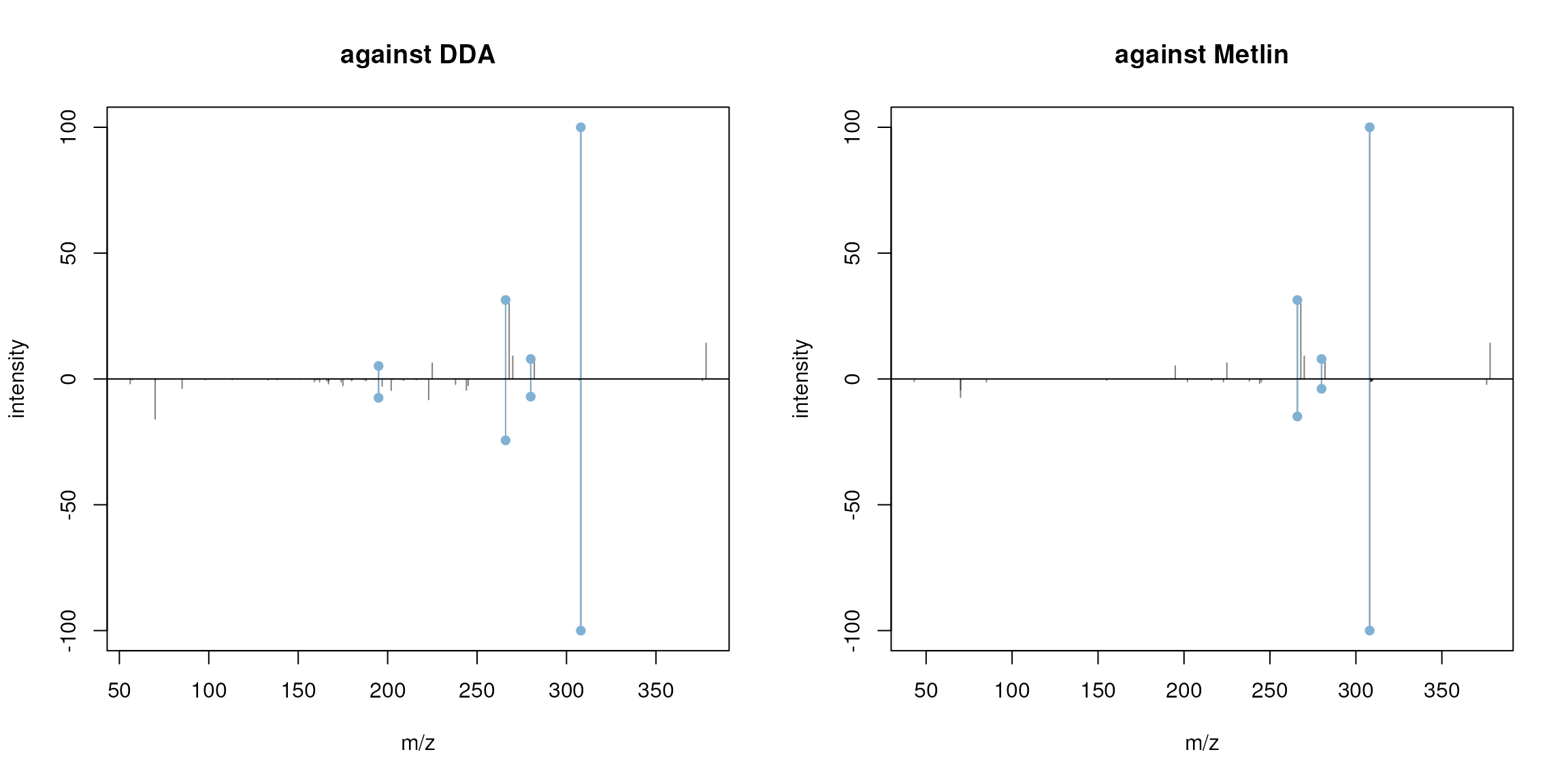
Mirror plot comparing the reconstructed MS2 spectrum for Prochloraz (upper panel) against the measured spectrum from the DDA data and the Prochloraz spectrum from Metlin.
The spectra fit relatively well. Interestingly, the peak representing the precursor (the right-most peak) seems to have a slightly shifted m/z value in the reconstructed spectrum. Also, by closer inspecting the spectrum two groups of peaks with small differences in m/z can be observed (see plot below).
plotSpectra(prochloraz_swath_spectrum)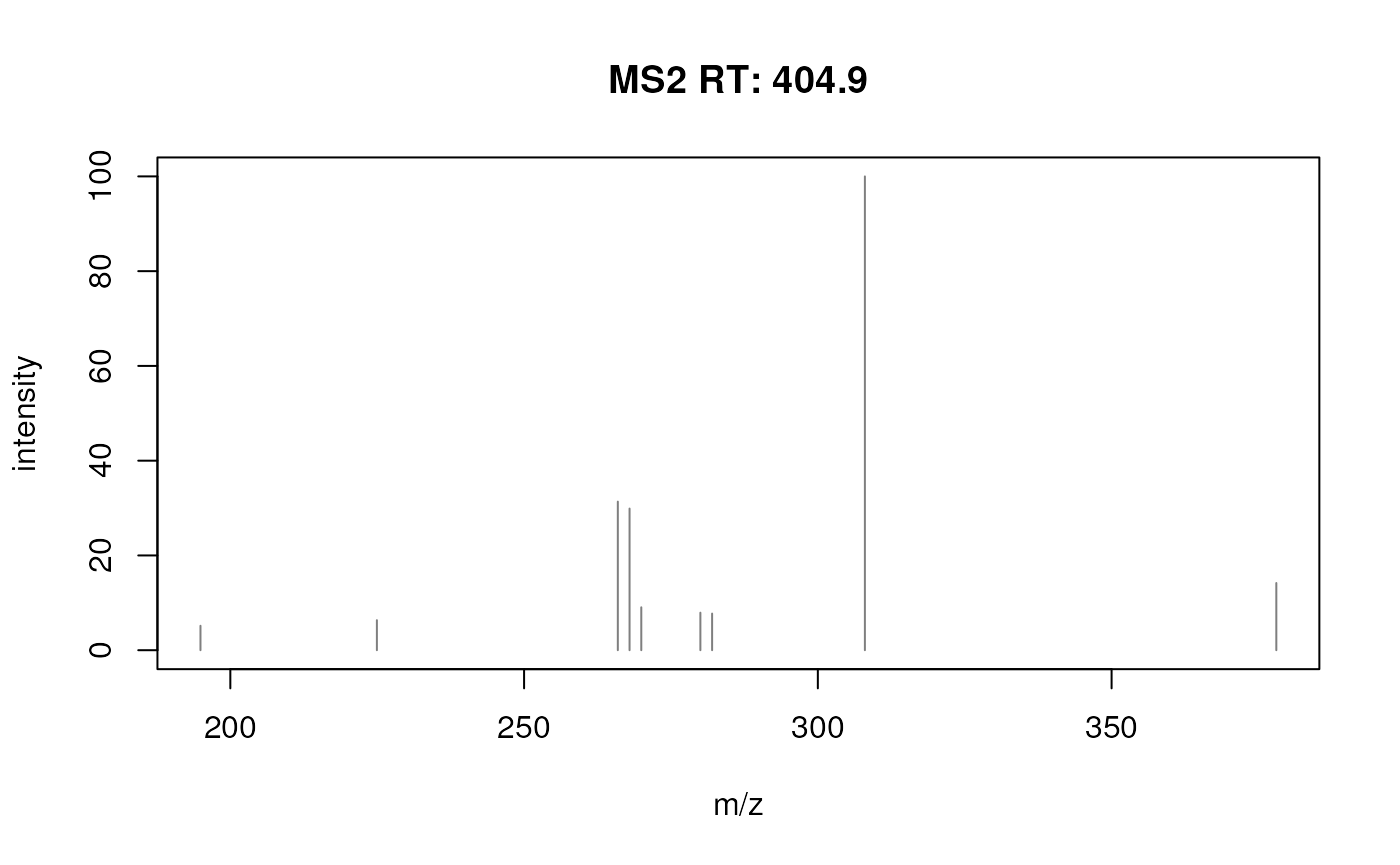
SWATH-derived MS2 spectrum for prochloraz.
These could represent fragments from isotopes of the original
compound. DIA MS2 data, since all ions at a given retention time are
fragmented, can contain fragments from isotopes. We thus below use the
isotopologues() function from the MetaboCoreUtils
package to check for presence of potential isotope peaks in the
reconstructed MS2 spectrum for prochloraz.
library(MetaboCoreUtils)
isotopologues(peaksData(prochloraz_swath_spectrum)[[1]])## [[1]]
## [1] 3 4 5
##
## [[2]]
## [1] 6 7Indeed, peaks 3, 4 and 5 as well as 6 and 7 have been assigned to a group of potential isotope peaks. While this is no proof that the peaks are indeed fragment isotopes of prochloraz it is highly likely (given their difference in m/z and relative intensity differences). Below we thus define a function that keeps only the monoisotopic peak for each isotope group in a spectrum.
## Function to keep only the first (monoisotopic) peak for potential
## isotopologue peak groups.
rem_iso <- function(x, ...) {
idx <- isotopologues(x)
idx <- unlist(lapply(idx, function(z) z[-1]), use.names = FALSE)
if (length(idx))
x[-idx, , drop = FALSE]
else x
}
prochloraz_swath_spectrum2 <- addProcessing(prochloraz_swath_spectrum,
rem_iso)
par(mfrow = c(1, 2))
plotSpectra(prochloraz_swath_spectrum)
plotSpectra(prochloraz_swath_spectrum2)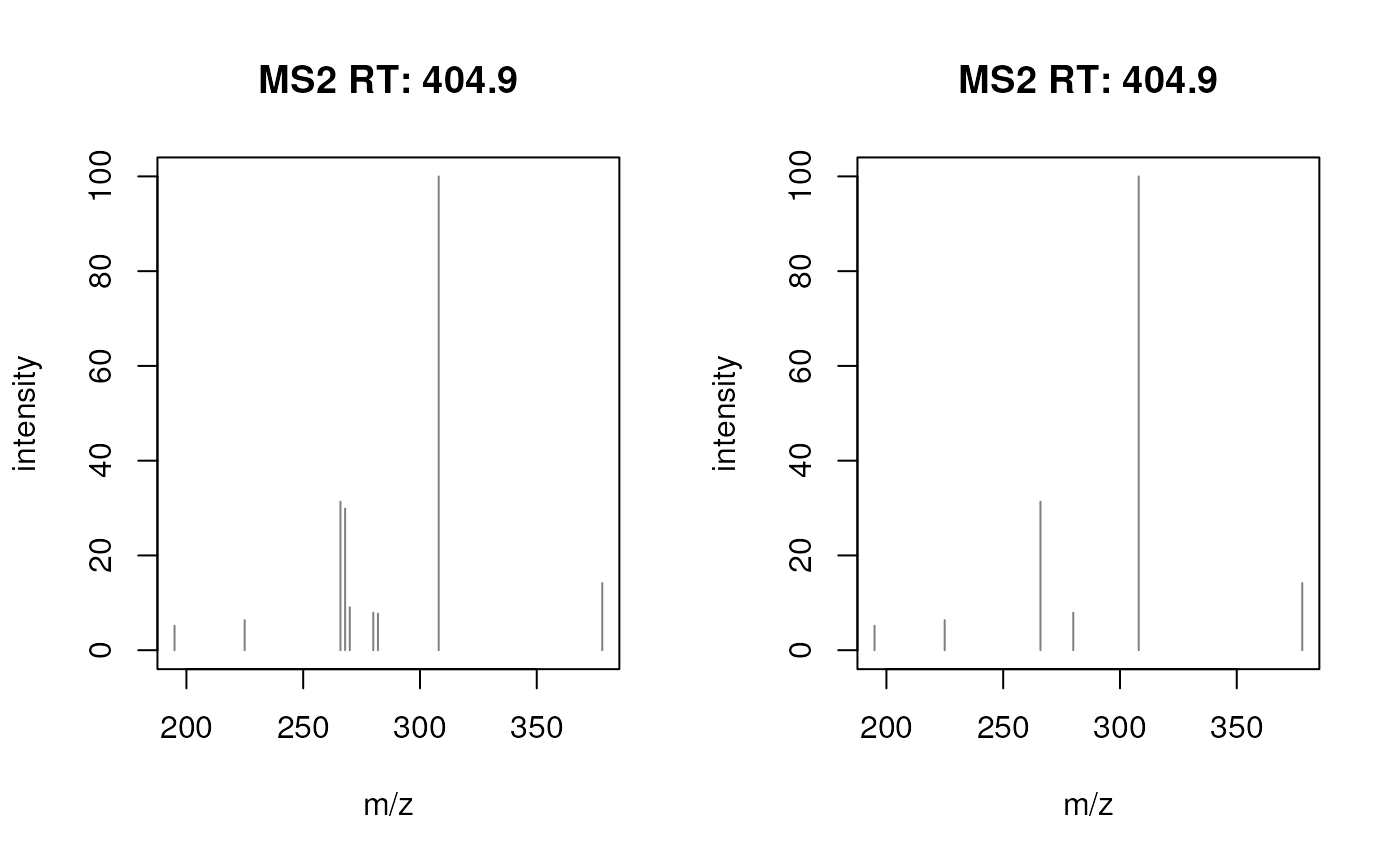
SWATH MS2 spectrum for prochloraz before (left) and after deisotoping (right).
Removing the isotope peaks from the SWATH MS2 spectrum increases also the spectra similarity score (since reference spectra generally will contain only fragments of the ion of interest, but not of any of its isotopes).
compareSpectra(prochloraz_swath_spectrum, prochloraz_dda_spectrum)## [1] 0.4623719
compareSpectra(prochloraz_swath_spectrum2, prochloraz_dda_spectrum)## [1] 0.5932303Similar to the DDA data, the reconstructed MS2 spectra from SWATH data could be used in the annotation of the MS1 chromatographic peaks.
Outlook
Currently, spectra data representation, handling and processing is being re-implemented as part of the RforMassSpectrometry initiative aiming at increasing the performance of methods and simplifying their use. Thus, parts of the workflow described here will be changed (improved) in future.
Along with these developments, improved matching strategies for
larger data sets will be implemented as well as functionality to compare
Spectra directly to reference MS2 spectra from public
annotation resources (e.g. Massbank or HMDB). Visit Metabonaut
or SpectraTutorials
for more examples and information.
Regarding SWATH data analysis, future development will involve improved selection of the correct MS2 chromatographic peaks considering also correlation with intensity values across several samples.
Session information
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] MetaboCoreUtils_1.20.1 MsBackendMgf_1.20.0 MsDataHub_1.12.0
## [4] MsExperiment_1.14.0 ProtGenerics_1.44.0 pander_0.6.6
## [7] Spectra_1.22.2 S4Vectors_0.50.1 BiocGenerics_0.58.1
## [10] generics_0.1.4 xcms_4.11.1 BiocParallel_1.46.0
## [13] BiocStyle_2.40.0
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0
## [3] MultiAssayExperiment_1.38.0 magrittr_2.0.5
## [5] farver_2.1.2 MALDIquant_1.22.3
## [7] rmarkdown_2.31 fs_2.1.0
## [9] ragg_1.5.2 vctrs_0.7.3
## [11] memoise_2.0.1 htmltools_0.5.9
## [13] S4Arrays_1.12.0 progress_1.2.3
## [15] AnnotationHub_4.2.2 curl_7.1.0
## [17] SparseArray_1.12.2 mzID_1.50.0
## [19] sass_0.4.10 bslib_0.11.0
## [21] htmlwidgets_1.6.4 desc_1.4.3
## [23] plyr_1.8.9 httr2_1.2.2
## [25] impute_1.86.0 cachem_1.1.0
## [27] igraph_2.3.2 lifecycle_1.0.5
## [29] iterators_1.0.14 pkgconfig_2.0.3
## [31] Matrix_1.7-5 R6_2.6.1
## [33] fastmap_1.2.0 MatrixGenerics_1.24.0
## [35] clue_0.3-68 digest_0.6.39
## [37] pcaMethods_2.4.0 AnnotationDbi_1.74.0
## [39] ExperimentHub_3.2.0 textshaping_1.0.5
## [41] GenomicRanges_1.64.0 RSQLite_3.53.2
## [43] filelock_1.0.3 httr_1.4.8
## [45] abind_1.4-8 compiler_4.6.0
## [47] withr_3.0.3 bit64_4.8.2
## [49] doParallel_1.0.17 S7_0.2.2
## [51] PTMods_1.0.0 DBI_1.3.0
## [53] MASS_7.3-65 rappdirs_0.3.4
## [55] DelayedArray_0.38.2 mzR_2.46.0
## [57] tools_4.6.0 PSMatch_1.16.0
## [59] otel_0.2.0 glue_1.8.1
## [61] QFeatures_1.22.0 grid_4.6.0
## [63] cluster_2.1.8.2 reshape2_1.4.5
## [65] gtable_0.3.6 preprocessCore_1.74.0
## [67] tidyr_1.3.2 data.table_1.18.4
## [69] hms_1.1.4 XVector_0.52.0
## [71] BiocVersion_3.23.1 foreach_1.5.2
## [73] pillar_1.11.1 stringr_1.6.0
## [75] limma_3.68.4 dplyr_1.2.1
## [77] BiocFileCache_3.2.0 lattice_0.22-9
## [79] bit_4.6.0 tidyselect_1.2.1
## [81] Biostrings_2.80.1 knitr_1.51
## [83] bookdown_0.47 IRanges_2.46.0
## [85] Seqinfo_1.2.0 SummarizedExperiment_1.42.0
## [87] xfun_0.59 Biobase_2.72.0
## [89] statmod_1.5.2 MSnbase_2.37.0
## [91] matrixStats_1.5.0 stringi_1.8.7
## [93] lazyeval_0.2.3 yaml_2.3.12
## [95] evaluate_1.0.5 codetools_0.2-20
## [97] MsCoreUtils_1.24.0 tibble_3.3.1
## [99] BiocManager_1.30.27 cli_3.6.6
## [101] affyio_1.82.0 systemfonts_1.3.2
## [103] jquerylib_0.1.4 Rcpp_1.1.1-1.1
## [105] MassSpecWavelet_1.78.0 dbplyr_2.6.0
## [107] png_0.1-9 XML_3.99-0.23
## [109] parallel_4.6.0 pkgdown_2.2.0.9000
## [111] ggplot2_4.0.3 blob_1.3.0
## [113] prettyunits_1.2.0 AnnotationFilter_1.36.0
## [115] MsFeatures_1.20.0 scales_1.4.0
## [117] affy_1.90.0 ncdf4_1.24
## [119] purrr_1.2.2 crayon_1.5.3
## [121] rlang_1.2.0 vsn_3.80.0
## [123] KEGGREST_1.52.2