chromatogram: extract chromatographic data (such as an extracted ion
chromatogram, a base peak chromatogram or total ion chromatogram) from
an MSnbase::OnDiskMSnExp or XCMSnExp objects. See also the help page of
the chromatogram function in the MSnbase package.
Usage
# S4 method for class 'XCMSnExp'
chromatogram(
object,
rt,
mz,
aggregationFun = "sum",
missing = NA_real_,
msLevel = 1L,
BPPARAM = bpparam(),
adjustedRtime = hasAdjustedRtime(object),
filled = FALSE,
include = c("apex_within", "any", "none"),
...
)Arguments
- object
Either a MSnbase::OnDiskMSnExp or XCMSnExp object from which the chromatograms should be extracted.
- rt
numeric(2)or two-columnmatrixdefining the lower and upper boundary for the retention time range(s). If not specified, the full retention time range of the original data will be used.- mz
numeric(2)or two-columnmatrixdefining the lower and upper mz value for the MS data slice(s). If not specified, the chromatograms will be calculated on the full mz range.- aggregationFun
character(1)specifying the function to be used to aggregate intensity values across the mz value range for the same retention time. Allowed values are"sum"(the default),"max","mean"and"min".- missing
numeric(1)allowing to specify the intensity value to be used if for a given retention time no signal was measured within the mz range of the corresponding scan. Defaults toNA_real_(see also Details and Notes sections below). Usemissing = 0to resemble the behaviour of thegetEICfrom the old user interface.- msLevel
integer(1)specifying the MS level from which the chromatogram should be extracted. Defaults tomsLevel = 1L.- BPPARAM
Parallelisation backend to be used, which will depend on the architecture. Default is
BiocParallel::bparam().- adjustedRtime
For
chromatogram,XCMSnExp: whether the adjusted (adjustedRtime = TRUE) or raw retention times (adjustedRtime = FALSE) should be used for filtering and returned in the resulting MSnbase::MChromatograms object. Adjusted retention times are used by default if available.- filled
logical(1)whether filled-in peaks should also be returned. Defaults tofilled = FALSE, i.e. returns only detected chromatographic peaks in the result object.- include
character(1)defining which chromatographic peaks should be returned. Supported areinclude = "apex_within"(the default) which returns chromatographic peaks that have their apex within themzrtrange,include = "any"to return all chromatographic peaks which m/z and rt ranges overlap themzandrtorinclude = "none"to not include any chromatographic peaks.- ...
optional parameters - currently ignored.
Value
chromatogram returns a XChromatograms object with
the number of columns corresponding to the number of files in
object and number of rows the number of specified ranges (i.e.
number of rows of matrices provided with arguments mz and/or
rt). All chromatographic peaks with their apex position within the
m/z and retention time range are also retained as well as all feature
definitions for these peaks.
Details
Arguments rt and mz allow to specify the MS data slice (i.e. the m/z
range and retention time window) from which the chromatogram should be
extracted. These parameters can be either a numeric of length 2 with the
lower and upper limit, or a matrix with two columns with the lower and
upper limits to extract multiple EICs at once.
The parameter aggregationSum allows to specify the function to be
used to aggregate the intensities across the m/z range for the same
retention time. Setting aggregationFun = "sum" would e.g. allow
to calculate the total ion chromatogram (TIC),
aggregationFun = "max" the base peak chromatogram (BPC).
If for a given retention time no intensity is measured in that spectrum a
NA intensity value is returned by default. This can be changed with the
parameter missing, setting missing = 0 would result in a 0 intensity
being returned in these cases.
Note
For XCMSnExp objects, if adjusted retention times are
available, the chromatogram method will by default report
and use these (for the subsetting based on the provided parameter
rt). This can be changed by setting adjustedRtime = FALSE.
See also
XCMSnExp for the data object.
MSnbase::Chromatogram() for the object representing chromatographic
data.
Examples
## Load a test data set with identified chromatographic peaks
library(MSnbase)
data(faahko_sub)
## Update the path to the files for the local system
dirname(faahko_sub) <- system.file("cdf/KO", package = "faahKO")
## Disable parallel processing for this example
register(SerialParam())
## Extract the ion chromatogram for one chromatographic peak in the data.
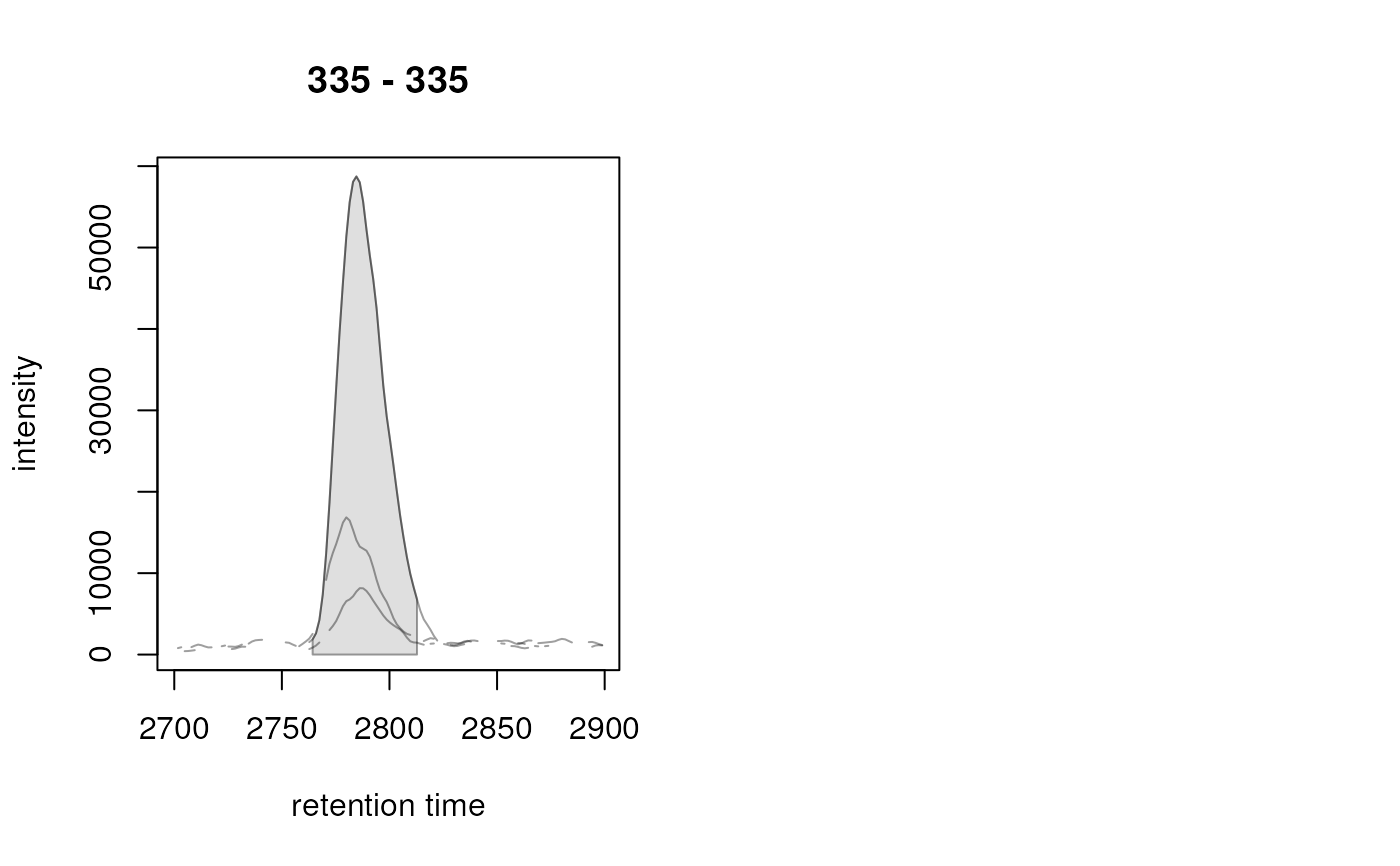
chrs <- chromatogram(faahko_sub, rt = c(2700, 2900), mz = 335)
chrs
#> XChromatograms with 1 row and 3 columns
#> ko15.CDF ko16.CDF ko18.CDF
#> <XChromatogram> <XChromatogram> <XChromatogram>
#> [1,] peaks: 0 peaks: 1 peaks: 0
#> phenoData with 1 variables
#> featureData with 5 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
## Identified chromatographic peaks
chromPeaks(chrs)
#> mz mzmin mzmax rt rtmin rtmax into intb maxo sn sample
#> CP095 335 335 335 2786.199 2764.29 2812.803 1496244 1451652 58736 55 2
#> row column
#> CP095 1 2
## Plot the chromatogram
plot(chrs)
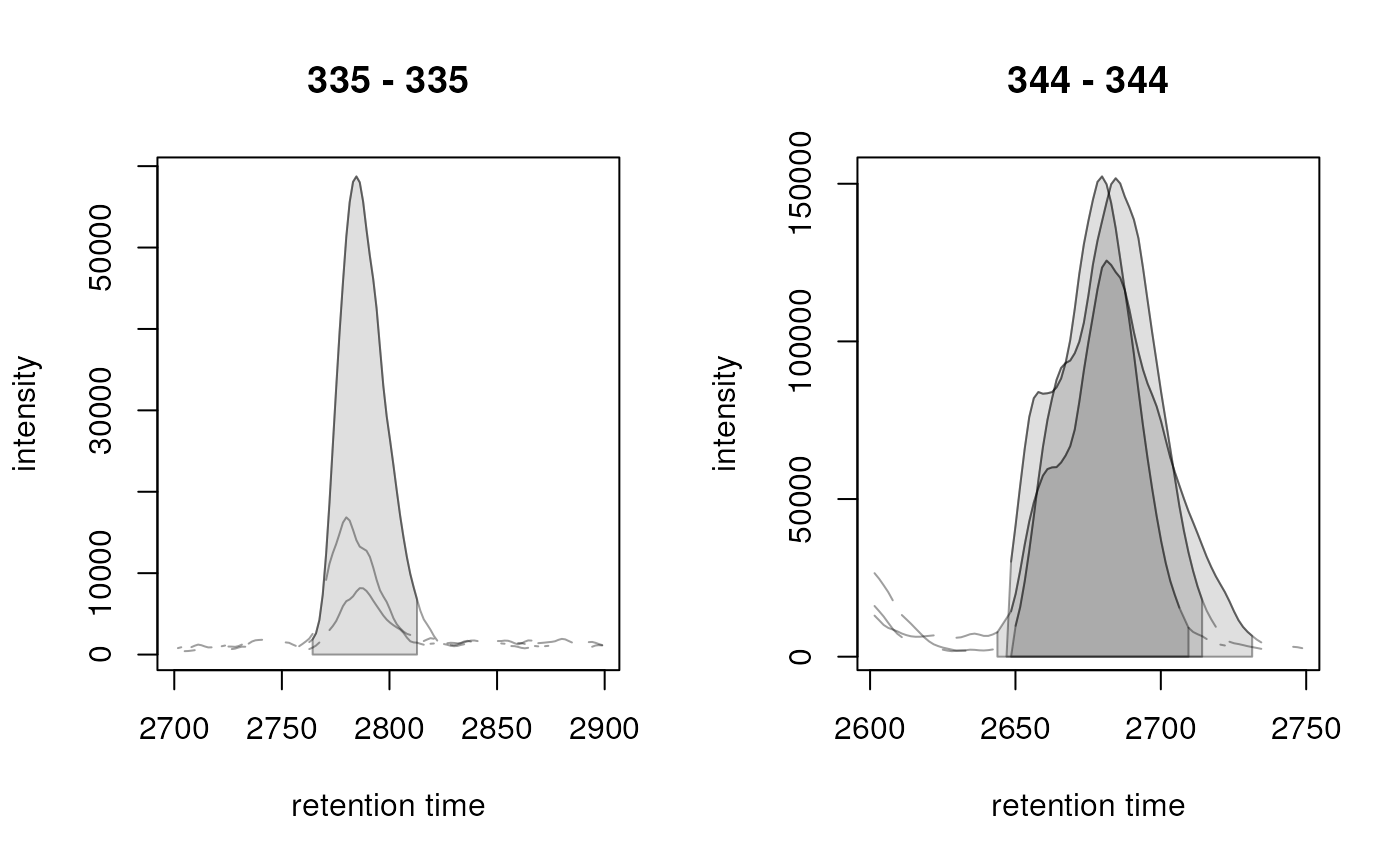
## Extract chromatograms for multiple ranges.
mzr <- matrix(c(335, 335, 344, 344), ncol = 2, byrow = TRUE)
rtr <- matrix(c(2700, 2900, 2600, 2750), ncol = 2, byrow = TRUE)
chrs <- chromatogram(faahko_sub, mz = mzr, rt = rtr)
chromPeaks(chrs)
#> mz mzmin mzmax rt rtmin rtmax into intb maxo sn
#> CP095 335 335 335 2786.199 2764.290 2812.803 1496244 1451652 58736 55
#> CP003 344 344 344 2679.783 2646.919 2709.517 5210016 5135917 152320 68
#> CP090 344 344 344 2686.042 2648.483 2714.211 5700652 5574745 151744 87
#> CP193 344 344 344 2682.914 2643.790 2731.427 5255689 4946143 125632 41
#> sample row column
#> CP095 2 1 2
#> CP003 1 2 1
#> CP090 2 2 2
#> CP193 3 2 3
plot(chrs)
 ## Get access to all chromatograms for the second mz/rt range
chrs[1, ]
#> XChromatograms with 1 row and 3 columns
#> ko15.CDF ko16.CDF ko18.CDF
#> <XChromatogram> <XChromatogram> <XChromatogram>
#> [1,] peaks: 0 peaks: 1 peaks: 0
#> phenoData with 1 variables
#> featureData with 5 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
## Plot just that one
plot(chrs[1, , drop = FALSE])
## Get access to all chromatograms for the second mz/rt range
chrs[1, ]
#> XChromatograms with 1 row and 3 columns
#> ko15.CDF ko16.CDF ko18.CDF
#> <XChromatogram> <XChromatogram> <XChromatogram>
#> [1,] peaks: 0 peaks: 1 peaks: 0
#> phenoData with 1 variables
#> featureData with 5 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
## Plot just that one
plot(chrs[1, , drop = FALSE])