Containers for chromatographic and peak detection data
Source:R/functions-XChromatograms.R, R/functions-XChromatogram.R, R/methods-XChromatogram.R, and 1 more
XChromatogram.RdThe XChromatogram object allows to store chromatographic data (e.g.
an extracted ion chromatogram) along with identified chromatographic peaks
within that data. The object inherits all functions from the
MSnbase::Chromatogram() object in the MSnbase package.
Multiple XChromatogram objects can be stored in a XChromatograms object.
This class extends MSnbase::MChromatograms() from the MSnbase package
and allows thus to arrange chromatograms in a matrix-like structure, columns
representing samples and rows m/z-retention time ranges.
All functions are described (grouped into topic-related sections) after the Arguments section.
Usage
XChromatograms(data, phenoData, featureData, chromPeaks, chromPeakData, ...)
XChromatogram(
rtime = numeric(),
intensity = numeric(),
mz = c(NA_real_, NA_real_),
filterMz = c(NA_real_, NA_real_),
precursorMz = c(NA_real_, NA_real_),
productMz = c(NA_real_, NA_real_),
fromFile = integer(),
aggregationFun = character(),
msLevel = 1L,
chromPeaks,
chromPeakData
)
# S4 method for class 'XChromatogram'
chromPeaks(
object,
rt = numeric(),
mz = numeric(),
ppm = 0,
type = c("any", "within", "apex_within"),
msLevel
)
# S4 method for class 'XChromatogram'
chromPeaks(object) <- value
# S4 method for class 'XChromatogram,ANY'
plot(
x,
col = "#00000060",
lty = 1,
type = "l",
xlab = "retention time",
ylab = "intensity",
main = NULL,
peakType = c("polygon", "point", "rectangle", "none"),
peakCol = "#00000060",
peakBg = "#00000020",
peakPch = 1,
...
)
# S4 method for class 'XChromatogram'
filterMz(object, mz, ...)
# S4 method for class 'XChromatogram'
filterRt(object, rt, ...)
# S4 method for class 'XChromatogram'
hasChromPeaks(object)
# S4 method for class 'XChromatogram'
dropFilledChromPeaks(object)
# S4 method for class 'XChromatogram'
chromPeakData(object)
# S4 method for class 'XChromatogram'
chromPeakData(object) <- value
# S4 method for class 'XChromatogram,MergeNeighboringPeaksParam'
refineChromPeaks(object, param = MergeNeighboringPeaksParam())
# S4 method for class 'XChromatogram'
filterChromPeaks(object, method = c("keepTop"), ...)
# S4 method for class 'XChromatogram'
transformIntensity(object, FUN = identity)
# S4 method for class 'XChromatograms'
hasChromPeaks(object)
# S4 method for class 'XChromatograms'
hasFilledChromPeaks(object)
# S4 method for class 'XChromatograms'
chromPeaks(
object,
rt = numeric(),
mz = numeric(),
ppm = 0,
type = c("any", "within", "apex_within"),
msLevel
)
# S4 method for class 'XChromatograms'
chromPeakData(object)
# S4 method for class 'XChromatograms'
filterMz(object, mz, ...)
# S4 method for class 'XChromatograms'
filterRt(object, rt, ...)
# S4 method for class 'XChromatograms,ANY'
plot(
x,
col = "#00000060",
lty = 1,
type = "l",
xlab = "retention time",
ylab = "intensity",
main = NULL,
peakType = c("polygon", "point", "rectangle", "none"),
peakCol = "#00000060",
peakBg = "#00000020",
peakPch = 1,
...
)
# S4 method for class 'XChromatograms'
processHistory(object, fileIndex, type)
# S4 method for class 'XChromatograms'
hasFeatures(object, ...)
# S4 method for class 'XChromatograms'
dropFeatureDefinitions(object, ...)
# S4 method for class 'XChromatograms,PeakDensityParam'
groupChromPeaks(object, param)
# S4 method for class 'XChromatograms'
featureDefinitions(
object,
mz = numeric(),
rt = numeric(),
ppm = 0,
type = c("any", "within", "apex_within")
)
# S4 method for class 'XChromatograms,ANY,ANY,ANY'
x[i, j, drop = TRUE]
# S4 method for class 'XChromatograms'
featureValues(
object,
method = c("medret", "maxint", "sum"),
value = "into",
intensity = "into",
missing = NA,
...
)
# S4 method for class 'XChromatograms'
plotChromPeakDensity(
object,
param,
col = "#00000060",
xlab = "retention time",
main = NULL,
peakType = c("polygon", "point", "rectangle", "none"),
peakCol = "#00000060",
peakBg = "#00000020",
peakPch = 1,
simulate = TRUE,
...
)
# S4 method for class 'XChromatograms'
dropFilledChromPeaks(object)
# S4 method for class 'XChromatograms,MergeNeighboringPeaksParam'
refineChromPeaks(object, param = MergeNeighboringPeaksParam())
# S4 method for class 'XChromatograms'
filterChromPeaks(object, method = c("keepTop"), ...)
# S4 method for class 'XChromatograms'
transformIntensity(object, FUN = identity)Arguments
- data
For
XChromatograms:listofChromatogramorXChromatogramobjects.- phenoData
For
XChromatograms: either adata.frame,AnnotatedDataFramedescribing the phenotypical information of the samples.- featureData
For
XChromatograms: either adata.frameorAnnotatedDataFramewith additional information for each row of chromatograms.- chromPeaks
For
XChromatogram:matrixwith required columns"rt","rtmin","rtmax","into","maxo"and"sn". ForXChromatograms:list, same length thandata, with the chromatographic peaks for each chromatogram. Each element has to be amatrix, the ordering has to match the order of the chromatograms indata.- chromPeakData
For
XChromatogram:DataFramewith optional additional annotations for each chromatographic peak. The number of rows has to match the number of chromatographic peaks.- ...
For
filterChromPeaks: additional parameters defining how to filter chromatographic peaks. See function description below for details.- rtime
For
XChromatogram:numericwith the retention times (length has to be equal to the length ofintensity).- intensity
For
XChromatogram:numericwith the intensity values (length has to be equal to the length ofrtime).- mz
For
XChromatogram:numeric(2)representing the m/z value range (min, max) on which the chromatogram was created. This is supposed to contain the real range of m/z values in contrast to thefilterMzbelow. ForchromPeaksandfeatureDefinitions:numeric(2)defining the m/z range for which chromatographic peaks or features should be returned. ForfilterMz:numeric(2)defining the m/z range for which chromatographic peaks should be retained.#'- filterMz
For
XChromatogram:numeric(2)representing the m/z value range (min, max) that was used to filter the original object on m/z dimension. If not applicable usefilterMz = c(0, 0).- precursorMz
For
XChromatogram:numeric(2)for SRM/MRM transitions. Represents the mz of the precursor ion. See details for more information.- productMz
For
XChromatogram:numeric(2)for SRM/MRM transitions. Represents the mz of the product. See details for more information.- fromFile
For
XChromatogram:integer(1)the index of the file within theOnDiskMSnExporMSnExpobject from which the chromatogram was extracted.- aggregationFun
For
XChromatogram:character(1)specifying the function that was used to aggregate intensity values for the same retention time across the m/z range.- msLevel
For
XChromatogram:integerwith the MS level from which the chromatogram was extracted. ForchromPeaksandchromPeakData: extract chromatographic peaks of a certain MS level.- object
An
XChromatogramorXChromatogramsobject.- rt
For
chromPeaksandfeatureDefinitions:numeric(2)defining the retention time range for which chromatographic peaks or features should be returned. ForfilterRt:numeric(2)defining the retention time range to reduceobjectto.- ppm
For
chromPeaksandfeatureDefinitions:numeric(1)defining a ppm to expand the provided m/z range.- type
For
chromPeaksandfeatureDefinitions:character(1)defining which peaks or features to return ifrtormzis provided:"any"(default) return all peaks that are even partially overlapping withrt,"within"return peaks that are completely withinrtand"apex_within"return peaks which apex is withinrt.For `plot`: what type of plot should be used for the chromatogram (such as `"l"` for lines, `"p"` for points etc), see help of [plot()] in the `graphics` package for more details. For `processHistory`: restrict returned processing steps to specific types. Use [processHistoryTypes()] to list all supported values.- value
For
chromPeaks<-: a numericmatrixwith required columns"rt","rtmin","rtmax","into"and"maxo".- x
For
plot: anXChromatogramorXChromatogramsobject.- col
For
plot: the color to be used to draw the chromatogram.- lty
For
plotandplotChromPeakDensity: the line type.- xlab
For
plotandplotChromPeakDensity: the x axis label.- ylab
For
plot: the y axis label.- main
For
plotandplotChromPeakDensity: an optional title for the plot.- peakType
For
plotandplotChromPeakDensity:character(1)defining how (and if) identified chromatographic peak within the chromatogram should be plotted. Options are"polygon"(default): draw the peak borders with thepeakColcolor and fill the peak area with thepeakBgcolor,"point": indicate the peak's apex with a point,"rectangle": draw a rectangle around the identified peak and"none": don't draw peaks.- peakCol
For
plotandplotChromPeakDensity: the foreground color for the peaks. ForpeakType = "polygon"andpeakType = "rectangle"this is the color for the border. UseNAto not use a foreground color. This should either be a single color or a vector of colors with the same length thanchromPeaks(x)has rows.- peakBg
For
plotandplotChromPeakDensity: the background color for the peaks. ForpeakType = "polygon"andpeakType = "rectangle"the peak are or rectangle will be filled with this color. UseNAto skip. This should be either a single color or a vector of colors with the same length thanchromPeaks(x)has rows.- peakPch
For
plotandplotChromPeakDensity: the point character to be used forpeakType = "point". Seeplot()in thegraphicspackage for more details.- param
For
groupChromPeaksandplotChromPeakDensity: aPeakDensityParam()object with the settings for the peak density correspondence analysis algorithm.- method
For
featureValues:character(1)specifying the method to resolve multi-peak mappings within the sample sample, i.e. to select the representative peak for a feature for which more than one peak was assigned in one sample. Options are"medret"(default): select the peak closest to the median retention time of the feature,"maxint": select the peak with the largest signal and"sum": sum the values of all peaks (only ifvalueis"into"or"maxo"). ForfilterChromPeaks:character(1)defining the method that should be used to filter chromatographic peaks. See help onfilterChromPeaksbelow for details.- FUN
For
transformIntensity: a function to transform the intensity values ofobject.- fileIndex
For
processHistory: optionalintegerspecifying the index of the files/samples for which the ProcessHistory objects should be returned.- i
For
[:integerwith the row indices to subset theXChromatogramsobject.- j
For
[:integerwith the column indices to subset theXChromatogramsobject.- drop
For
[:logical(1)whether the dimensionality should be dropped (if possible). Defaults todrop = TRUE, thus, if length ofiandjis 1 aXChromatogramis returned. Note thatdropis ignored if length ofiorjis larger than 1, thus aXChromatogramsis returned.- missing
For
featureValues: how missing values should be reported. Allowed values areNA(default), anumeric(1)to replaceNAs with that value ormissing = "rowmin_half"to replaceNAs with half of the row's minimal (non-missing) value.- simulate
For
plotChromPeakDensity:logical(1)whether a correspondence analysis should be simulated based on the available data and the providedPeakDensityParam()paramargument. See section Correspondence analysis for details.
Note
Highlighting the peak area(s) in an XChromatogram or XChromatograms
object (plot with peakType = "polygon") draws a polygon representing
the displayed chromatogram from the peak's minimal retention time to the
maximal retention time. If the XChromatograms was extracted from an
XCMSnExp() object with the chromatogram() function this might not
represent the actual identified peak area if the m/z range that was
used to extract the chromatogram was larger than the peak's m/z.
Creation of objects
Objects can be created with the contructor function XChromatogram and
XChromatograms, respectively. Also, they can be coerced from
MSnbase::Chromatogram() or MSnbase::MChromatograms() objects using
as(object, "XChromatogram") or as(object, "XChromatograms").
Filtering and subsetting
Besides classical subsetting with [ specific filter operations on
MSnbase::MChromatograms() and XChromatograms objects are available. See
filterColumnsIntensityAbove() for more details.
[allows to subset aXChromatogramsobject by row (i) and column (j), withiandjbeing of typeinteger. ThefeatureDefinitionswill also be subsetted accordingly and thepeakidxcolumn updated.filterMzfilters the chromatographic peaks within anXChromatogramorXChromatograms, if a column"mz"is present in thechromPeaksmatrix. This would be the case if theXChromatogramwas extracted from anXCMSnExp()object with thechromatogram()function. All chromatographic peaks with their m/z within the m/z range defined bymzwill be retained. Also feature definitions (if present) will be subset accordingly. The function returns a filteredXChromatogramorXChromatogramsobject.filterRtfilters chromatogram(s) by the provided retention time range. All eventually present chromatographic peaks with their apex within the retention time range specified withrtwill be retained. Also feature definitions, if present, will be filtered accordingly. The function returns a filteredXChromatogramorXChromatogramsobject.
Accessing data
See also help of MSnbase::Chromatogram() in the MSnbase package for
general information and data access. The methods listed here are specific for
XChromatogram and XChromatograms objects.
chromPeaks,chromPeaks<-: extract or set the matrix with the chromatographic peak definitions. Parameterrtallows to specify a retention time range for which peaks should be returned along with parametertypethat defines how overlapping is defined (parameter description for details). ForXChromatogramobjects the function returns amatrixwith columns"mz"(mean m/z value),"mzmin"(minimal m/z value) and"mzmax"(maximal m/z value),"rt"(retention time of the peak apex),"rtmin"(the lower peak boundary in retention time dimension),"rtmax"(the upper peak boundary in retention time dimension),"into"(the integrated peak signal/area of the peak),"maxo"(the maximum instensity of the peak and"sn"(the signal to noise ratio). Note that, depending on the peak detection algorithm, the matrix may contain additional columns. ForXChromatogramsobjects thematrixcontains also columns"row"and"column"specifying in which chromatogram ofobjectthe peak was identified. Chromatographic peaks are ordered by row.chromPeakData,chromPeakData<-: extract or set theS4Vectors::DataFrame()with optional chromatographic peak annotations.hasChromPeaks: infer whether aXChromatogram(orXChromatograms) has chromatographic peaks. ForXChromatogram: returns alogical(1), forXChromatograms: returns amatrix, same dimensions thanobjectwith eitherTRUEorFALSEif chromatographic peaks are available in the chromatogram at the respective position.hasFilledChromPeaks: whether aXChromatogram(or aXChromatogramin aXChromatograms) has filled-in chromatographic peaks. ForXChromatogram: returns alogical(1), forXChromatograms: returns amatrix, same dimensions thanobjectwith eitherTRUEorFALSEif chromatographic peaks are available in the chromatogram at the respective position.dropFilledChromPeaks: removes filled-in chromatographic peaks. SeedropFilledChromPeaks()help forXCMSnExp()objects for more information.hasFeatures: forXChromatogramsobjects only: if correspondence analysis has been performed and m/z-rt feature definitions are present. Returns alogical(1).dropFeatureDefinitions: forXChrmomatogramsobjects only: delete any correspondence analysis results (and related process history).featureDefinitions: forXChromatogramsobjects only. Extract the results from the correspondence analysis (performed withgroupChromPeaks). Returns aDataFramewith the properties of the defined m/z-rt features: their m/z and retention time range. Columnpeakidxcontains the index of the chromatographic peaks in thechromPeaksmatrix associated with the feature. Column"row"contains the row in theXChromatogramsobject in which the feature was defined. Similar to thechromPeaksmethod it is possible to filter the returned feature matrix with themz,rtandppmparameters.featureValues: forXChromatogramsobjects only. Extract the abundance estimates for the individuals features. Note that by default (with parametervalue = "index"amatrixof indices of the peaks in thechromPeaksmatrix associated to the feature is returned. To extract the integrated peak area usevalue = "into". The function returns amatrixwith one row per feature (infeatureDefinitions) and each column being a sample (i.e. column ofobject). For features without a peak associated in a certain sampleNAis returned. This can be changed with themissingargument of the function.filterChromPeaks: filters chromatographic peaks inobjectdepending on parametermethodand method-specific parameters passed as additional arguments with.... Available methods are:method = "keepTop": keep topn(defaultn = 1L) peaks in each chromatogram ordered by columnorder(defaults toorder = "maxo"). Parameterdecreasing(defaultdecreasing = TRUE) can be used to order peaks in descending (decreasing = TRUE) or ascending (decreasing = FALSE) order to keep the topnpeaks with largest or smallest values, respectively.
processHistory: returns alistof ProcessHistory objects representing the individual performed processing steps. Optional parameterstypeandfileIndexallow to further specify which processing steps to return.
Manipulating data
transformIntensity: transforms the intensity values of the chromatograms with provided functionFUN. SeeMSnbase::transformIntensity()in the MSnbase package for details. ForXChromatogramandXChromatogramsin addition to the intensity values also columns"into"and"maxo"in the object'schromPeaksmatrix are transformed by the same function.
Plotting and visualizing
plotdraws the chromatogram and highlights in addition any chromatographic peaks present in theXChromatogramorXChromatograms(unlesspeakType = "none"was specified). To draw peaks in different colors a vector of color definitions with length equal tonrow(chromPeaks(x))has to be submitted withpeakColand/orpeakBgdefining one color for each peak (in the order as peaks are inchromPeaks(x)). For base peak chromatograms or total ion chromatograms it might be better to setpeakType = "none"to avoid generating busy plots.plotChromPeakDensity: visualize peak density-based correspondence analysis results. See section Correspondence analysis for more details.
Chromatographic peak detection
See findChromPeaks-Chromatogram-CentWaveParam for information.
After chromatographic peak detection it is also possible to refine
identified chromatographic peaks with the refineChromPeaks method (e.g. to
reduce peak detection artifacts). Currently, only peak refinement using the
merge neighboring peaks method is available (see
MergeNeighboringPeaksParam() for a detailed description of the approach.
Correspondence analysis
Identified chromatographic peaks in an XChromatograms object can be grouped
into features with the groupChromPeaks function. Currently, such a
correspondence analysis can be performed with the peak density method
(see groupChromPeaks for more details) specifying the algorithm settings
with a PeakDensityParam() object. A correspondence analysis is performed
separately for each row in the XChromatograms object grouping
chromatographic peaks across samples (columns).
The analysis results are stored in the returned XChromatograms object
and can be accessed with the featureDefinitions method which returns a
DataFrame with one row for each feature. Column "row" specifies in
which row of the XChromatograms object the feature was identified.
The plotChromPeakDensity method can be used to visualize peak density
correspondence results, or to simulate a peak density correspondence
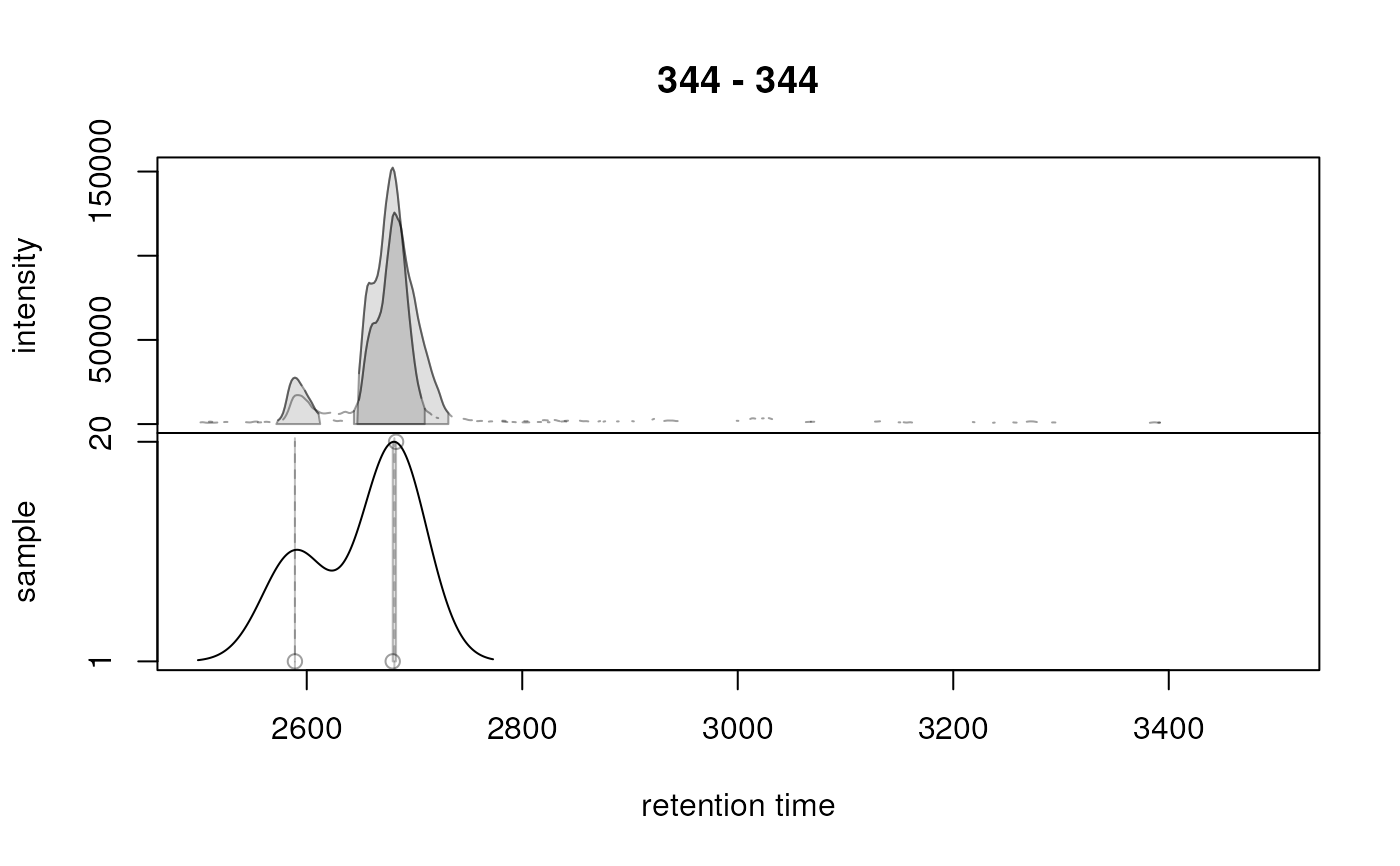
analysis on chromatographic data. The resulting plot consists of two panels,
the upper panel showing the chromatographic data as well as the identified
chromatographic peaks, the lower panel the distribution of peaks (the peak
density) along the retention time axis. This plot shows each peak as a point
with it's peak's retention time on the x-axis, and the sample in which it
was found on the y-axis. The distribution of peaks along the retention time
axis is visualized with a density estimate. Grouped chromatographic peaks
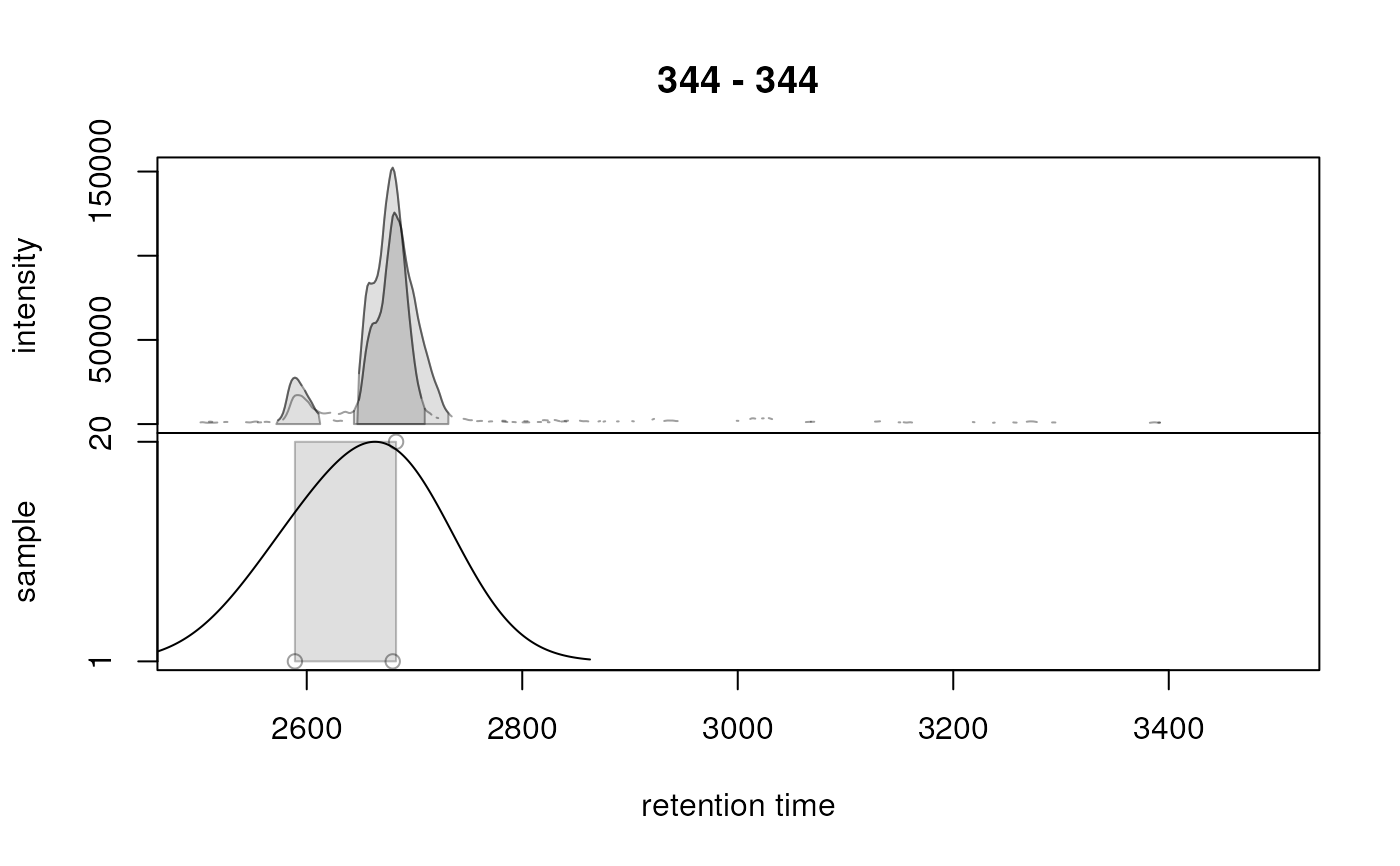
are indicated with grey shaded rectangles. Parameter simulate allows to
define whether the correspondence analysis should be simulated (
simulate=TRUE, based on the available data and the provided
PeakDensityParam() parameter class) or not (simulate=FALSE). For the
latter it is assumed that a correspondence analysis has been performed with
the peak density method on the object.
See examples below.
Abundance estimates for each feature can be extracted with the
featureValues function using parameter value = "into" to extract the
integrated peak area for each feature. The result is a matrix, columns
being samples and rows features.
See also
findChromPeaks-centWave for peak
detection on MSnbase::MChromatograms() objects.
Examples
## ---- Creation of XChromatograms ----
##
## Create a XChromatograms from Chromatogram objects
library(MSnbase)
dta <- list(Chromatogram(rtime = 1:7, c(3, 4, 6, 12, 8, 3, 2)),
Chromatogram(1:10, c(4, 6, 3, 4, 7, 13, 43, 34, 23, 9)))
## Create an XChromatograms without peak data
xchrs <- XChromatograms(dta)
## Create an XChromatograms with peaks data
pks <- list(matrix(c(4, 2, 5, 30, 12, NA), nrow = 1,
dimnames = list(NULL, c("rt", "rtmin", "rtmax", "into", "maxo", "sn"))),
NULL)
xchrs <- XChromatograms(dta, chromPeaks = pks)
## Create an XChromatograms from XChromatogram objects
dta <- lapply(dta, as, "XChromatogram")
chromPeaks(dta[[1]]) <- pks[[1]]
xchrs <- XChromatograms(dta, nrow = 1)
hasChromPeaks(xchrs)
#> 1 2
#> 1 TRUE FALSE
## Loading a test data set with identified chromatographic peaks
faahko_sub <- loadXcmsData("faahko_sub2")
## Subset the dataset to the first and third file.
xod_sub <- filterFile(faahko_sub, file = c(1, 3))
od <- as(xod_sub, "MsExperiment")
## Extract chromatograms for a m/z - retention time slice
chrs <- chromatogram(od, mz = 344, rt = c(2500, 3500))
chrs
#> MChromatograms with 1 row and 2 columns
#> 1 2
#> <Chromatogram> <Chromatogram>
#> [1,] length: 639 length: 639
#> phenoData with 2 variables
#> featureData with 4 variables
## --------------------------------------------------- ##
## Chromatographic peak detection ##
## --------------------------------------------------- ##
## Perform peak detection using CentWave
xchrs <- findChromPeaks(chrs, param = CentWaveParam())
xchrs
#> XChromatograms with 1 row and 2 columns
#> 1 2
#> <XChromatogram> <XChromatogram>
#> [1,] peaks: 2 peaks: 1
#> phenoData with 2 variables
#> featureData with 4 variables
#> - - - xcms preprocessing - - -
#> Chromatographic peak detection:
#> method: centWave
## Do we have chromatographic peaks?
hasChromPeaks(xchrs)
#> 1 2
#> 1 TRUE TRUE
## Process history
processHistory(xchrs)
#> [[1]]
#> Object of class "XProcessHistory"
#> type: Peak detection
#> date: Sun Jul 5 13:00:15 2026
#> info:
#> fileIndex: 1,2
#> Parameter class: CentWaveParam
#>
## The chromatographic peaks, columns "row" and "column" provide information
## in which sample the peak was identified.
chromPeaks(xchrs)
#> mz mzmin mzmax rt rtmin rtmax into intb maxo sn
#> mzmin 344 344 344 2589.015 2571.801 2612.490 592297.3 554533.9 27600 14
#> mzmin 344 344 344 2679.783 2646.919 2709.517 5210015.9 5071346.6 152320 30
#> mzmin 344 344 344 2682.914 2643.790 2731.427 5255689.5 4983388.9 125632 28
#> row column
#> mzmin 1 1
#> mzmin 1 1
#> mzmin 1 2
## Spectifically extract chromatographic peaks for one sample/chromatogram
chromPeaks(xchrs[1, 2])
#> mz mzmin mzmax rt rtmin rtmax into intb maxo sn
#> mzmin 344 344 344 2682.914 2643.79 2731.427 5255689 4983389 125632 28
## Plot the results
plot(xchrs)
 ## Plot the results using a different color for each sample
sample_colors <- c("#ff000040", "#00ff0040", "#0000ff40")
cols <- sample_colors[chromPeaks(xchrs)[, "column"]]
plot(xchrs, col = sample_colors, peakBg = cols)
## Plot the results using a different color for each sample
sample_colors <- c("#ff000040", "#00ff0040", "#0000ff40")
cols <- sample_colors[chromPeaks(xchrs)[, "column"]]
plot(xchrs, col = sample_colors, peakBg = cols)
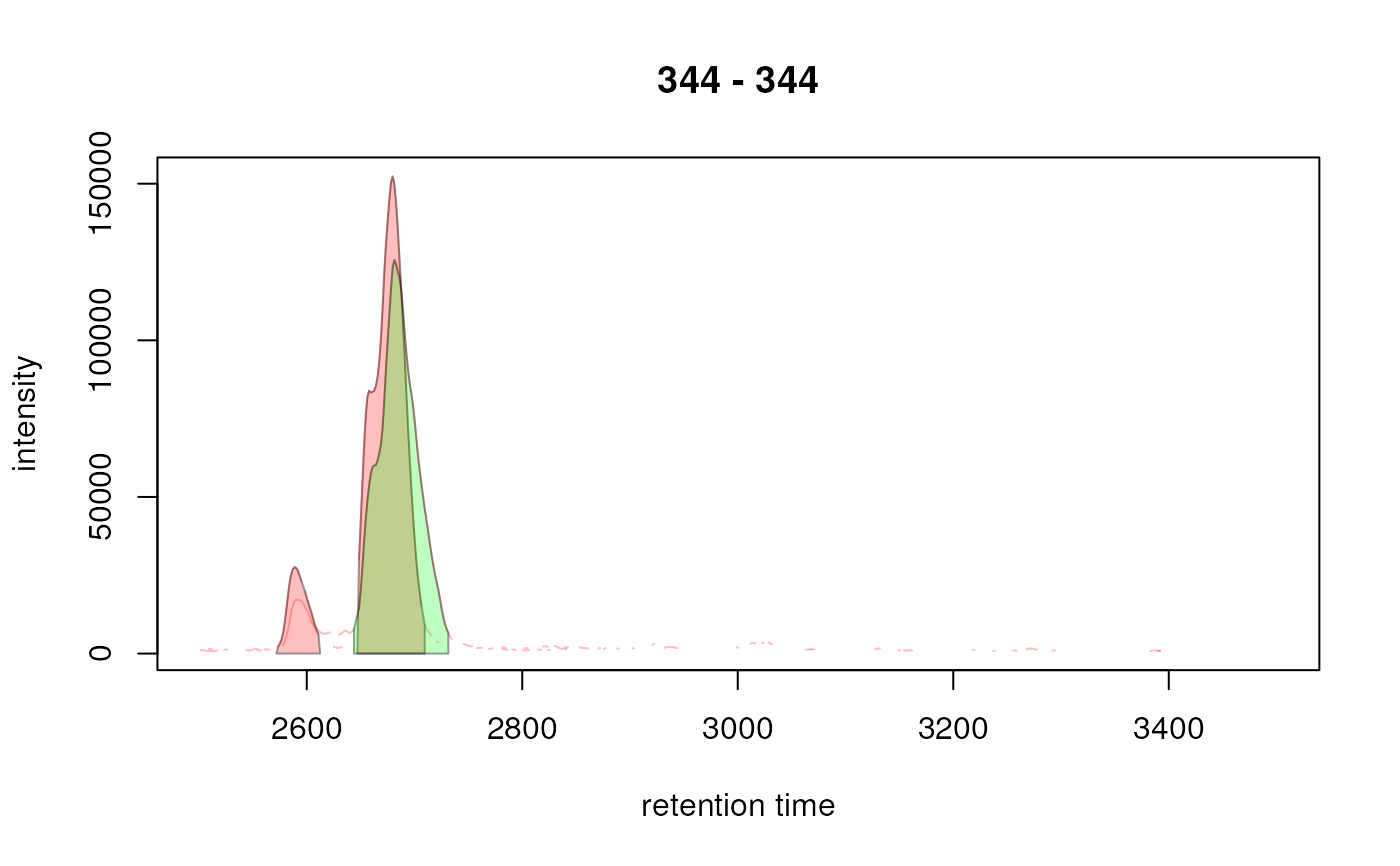 ## Indicate the peaks with a rectangle
plot(xchrs, col = sample_colors, peakCol = cols, peakType = "rectangle",
peakBg = NA)
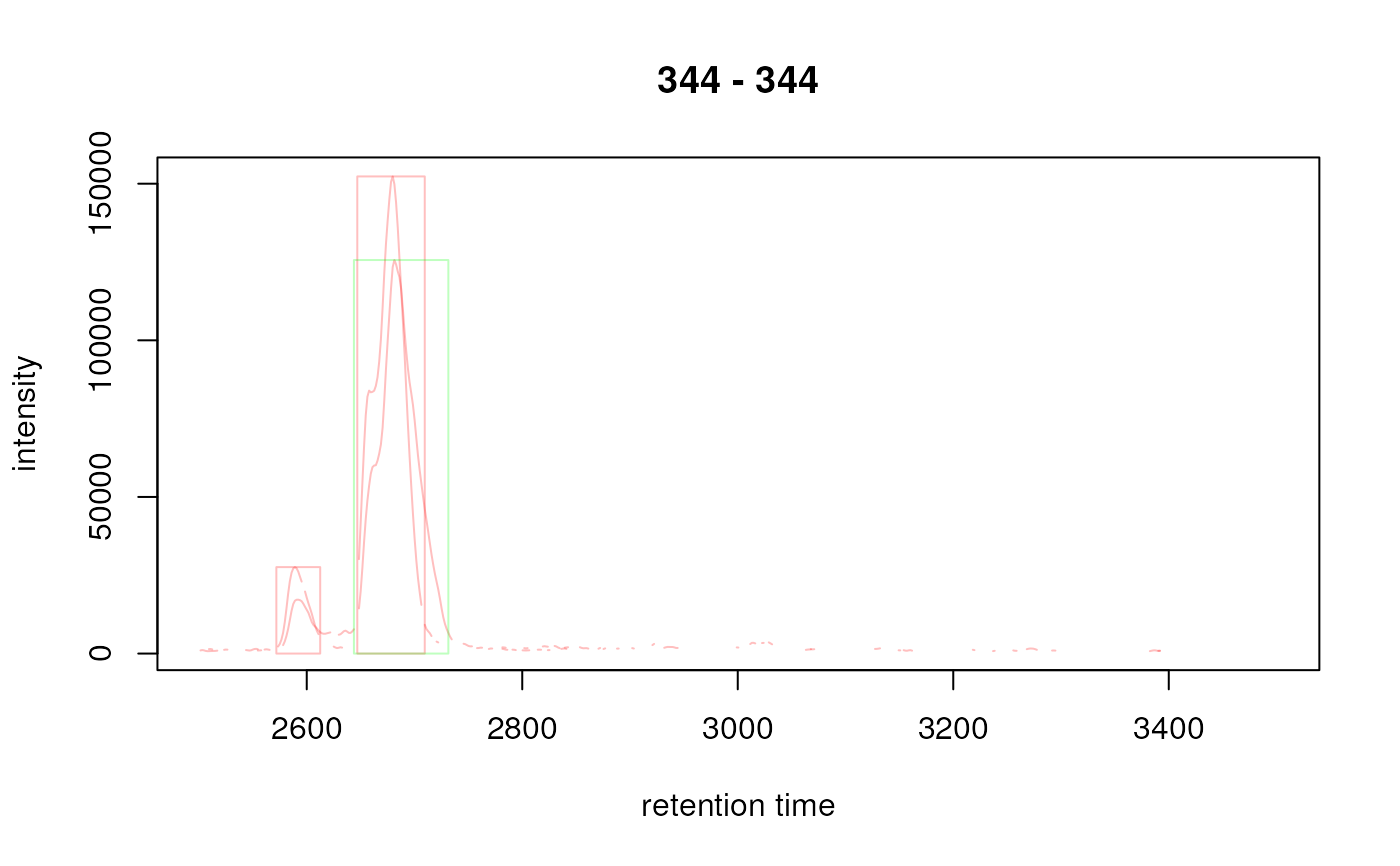
## Indicate the peaks with a rectangle
plot(xchrs, col = sample_colors, peakCol = cols, peakType = "rectangle",
peakBg = NA)
 ## --------------------------------------------------- ##
## Correspondence analysis ##
## --------------------------------------------------- ##
## Group chromatographic peaks across samples
prm <- PeakDensityParam(sampleGroup = rep(1, 2))
res <- groupChromPeaks(xchrs, param = prm)
hasFeatures(res)
#> [1] TRUE
featureDefinitions(res)
#> DataFrame with 2 rows and 10 columns
#> mzmed mzmin mzmax rtmed rtmin rtmax npeaks
#> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
#> FT1 344 344 344 2681.35 2679.78 2682.91 2
#> FT2 344 344 344 2589.01 2589.01 2589.01 1
#> X1 peakidx row
#> <numeric> <list> <integer>
#> FT1 2 2,3 1
#> FT2 1 1 1
## Plot the correspondence results. Use simulate = FALSE to show the
## actual results. Grouped chromatographic peaks are indicated with
## grey shaded rectangles.
plotChromPeakDensity(res, simulate = FALSE)
## --------------------------------------------------- ##
## Correspondence analysis ##
## --------------------------------------------------- ##
## Group chromatographic peaks across samples
prm <- PeakDensityParam(sampleGroup = rep(1, 2))
res <- groupChromPeaks(xchrs, param = prm)
hasFeatures(res)
#> [1] TRUE
featureDefinitions(res)
#> DataFrame with 2 rows and 10 columns
#> mzmed mzmin mzmax rtmed rtmin rtmax npeaks
#> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
#> FT1 344 344 344 2681.35 2679.78 2682.91 2
#> FT2 344 344 344 2589.01 2589.01 2589.01 1
#> X1 peakidx row
#> <numeric> <list> <integer>
#> FT1 2 2,3 1
#> FT2 1 1 1
## Plot the correspondence results. Use simulate = FALSE to show the
## actual results. Grouped chromatographic peaks are indicated with
## grey shaded rectangles.
plotChromPeakDensity(res, simulate = FALSE)
 ## Simulate a correspondence analysis based on different settings. Larger
## bw will increase the smoothing of the density estimate hence grouping
## chromatographic peaks that are more apart on the retention time axis.
prm <- PeakDensityParam(sampleGroup = rep(1, 3), bw = 60)
plotChromPeakDensity(res, param = prm)
## Simulate a correspondence analysis based on different settings. Larger
## bw will increase the smoothing of the density estimate hence grouping
## chromatographic peaks that are more apart on the retention time axis.
prm <- PeakDensityParam(sampleGroup = rep(1, 3), bw = 60)
plotChromPeakDensity(res, param = prm)
 ## Delete the identified feature definitions
res <- dropFeatureDefinitions(res)
hasFeatures(res)
#> [1] FALSE
library(MSnbase)
## Create a XChromatogram object
pks <- matrix(nrow = 1, ncol = 6)
colnames(pks) <- c("rt", "rtmin", "rtmax", "into", "maxo", "sn")
pks[, "rtmin"] <- 2
pks[, "rtmax"] <- 9
pks[, "rt"] <- 4
pks[, "maxo"] <- 19
pks[, "into"] <- 93
xchr <- XChromatogram(rtime = 1:10,
intensity = c(4, 8, 14, 19, 18, 12, 9, 8, 5, 2),
chromPeaks = pks)
xchr
#> Object of class: XChromatogram
#> length of object: 10
#> from file:
#> mz range: [NA, NA]
#> rt range: [1, 10]
#> MS level: 1
#> Identified chromatographic peaks (1):
#> rt rtmin rtmax into maxo sn
#> 4 2 9 93 19 NA
## Add arbitrary peak annotations
df <- DataFrame(peak_id = c("a"))
xchr <- XChromatogram(rtime = 1:10,
intensity = c(4, 8, 14, 19, 18, 12, 9, 8, 5, 2),
chromPeaks = pks, chromPeakData = df)
xchr
#> Object of class: XChromatogram
#> length of object: 10
#> from file:
#> mz range: [NA, NA]
#> rt range: [1, 10]
#> MS level: 1
#> Identified chromatographic peaks (1):
#> rt rtmin rtmax into maxo sn
#> 4 2 9 93 19 NA
chromPeakData(xchr)
#> DataFrame with 1 row and 3 columns
#> peak_id ms_level is_filled
#> <character> <integer> <logical>
#> 1 a 1 FALSE
## Extract the chromatographic peaks
chromPeaks(xchr)
#> rt rtmin rtmax into maxo sn
#> [1,] 4 2 9 93 19 NA
## Plotting of a single XChromatogram object
## o Don't highlight chromatographic peaks
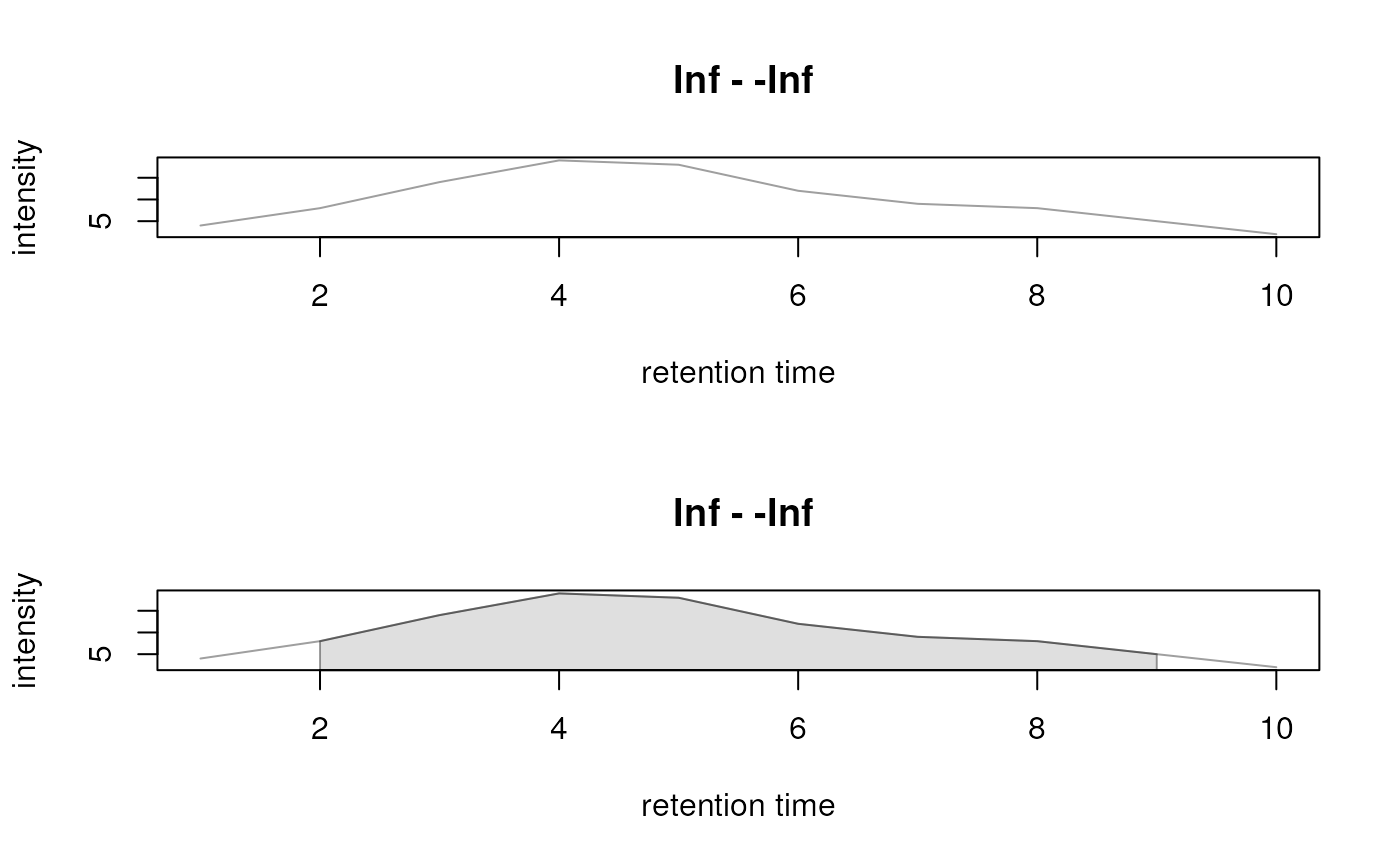
plot(xchr, peakType = "none")
## o Indicate peaks with a polygon
plot(xchr)
## Delete the identified feature definitions
res <- dropFeatureDefinitions(res)
hasFeatures(res)
#> [1] FALSE
library(MSnbase)
## Create a XChromatogram object
pks <- matrix(nrow = 1, ncol = 6)
colnames(pks) <- c("rt", "rtmin", "rtmax", "into", "maxo", "sn")
pks[, "rtmin"] <- 2
pks[, "rtmax"] <- 9
pks[, "rt"] <- 4
pks[, "maxo"] <- 19
pks[, "into"] <- 93
xchr <- XChromatogram(rtime = 1:10,
intensity = c(4, 8, 14, 19, 18, 12, 9, 8, 5, 2),
chromPeaks = pks)
xchr
#> Object of class: XChromatogram
#> length of object: 10
#> from file:
#> mz range: [NA, NA]
#> rt range: [1, 10]
#> MS level: 1
#> Identified chromatographic peaks (1):
#> rt rtmin rtmax into maxo sn
#> 4 2 9 93 19 NA
## Add arbitrary peak annotations
df <- DataFrame(peak_id = c("a"))
xchr <- XChromatogram(rtime = 1:10,
intensity = c(4, 8, 14, 19, 18, 12, 9, 8, 5, 2),
chromPeaks = pks, chromPeakData = df)
xchr
#> Object of class: XChromatogram
#> length of object: 10
#> from file:
#> mz range: [NA, NA]
#> rt range: [1, 10]
#> MS level: 1
#> Identified chromatographic peaks (1):
#> rt rtmin rtmax into maxo sn
#> 4 2 9 93 19 NA
chromPeakData(xchr)
#> DataFrame with 1 row and 3 columns
#> peak_id ms_level is_filled
#> <character> <integer> <logical>
#> 1 a 1 FALSE
## Extract the chromatographic peaks
chromPeaks(xchr)
#> rt rtmin rtmax into maxo sn
#> [1,] 4 2 9 93 19 NA
## Plotting of a single XChromatogram object
## o Don't highlight chromatographic peaks
plot(xchr, peakType = "none")
## o Indicate peaks with a polygon
plot(xchr)
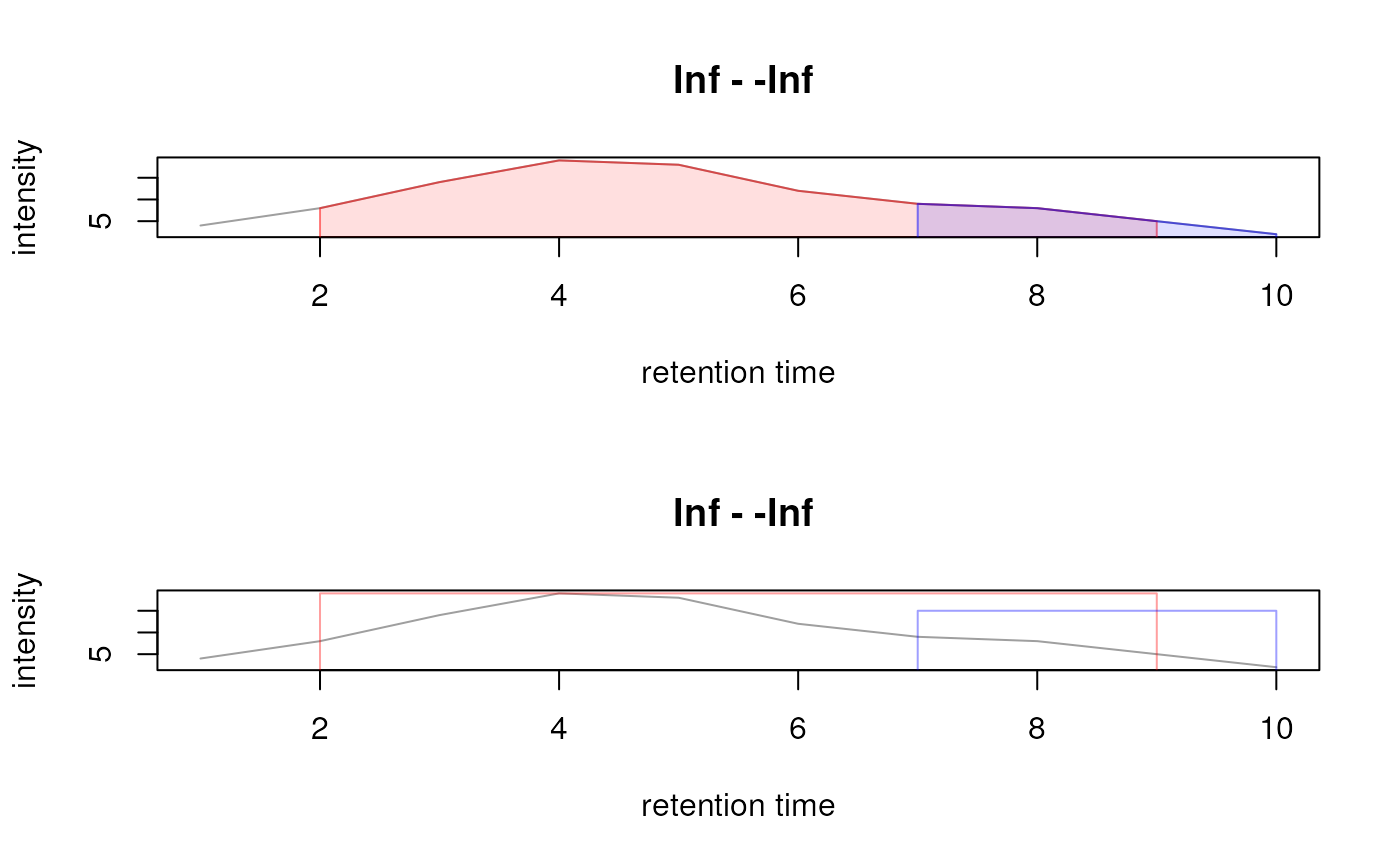
 ## Add a second peak to the data.
pks <- rbind(chromPeaks(xchr), c(7, 7, 10, NA, 15, NA))
chromPeaks(xchr) <- pks
## Plot the peaks in different colors
plot(xchr, peakCol = c("#ff000080", "#0000ff80"),
peakBg = c("#ff000020", "#0000ff20"))
## Indicate the peaks as rectangles
plot(xchr, peakCol = c("#ff000060", "#0000ff60"), peakBg = NA,
peakType = "rectangle")
## Add a second peak to the data.
pks <- rbind(chromPeaks(xchr), c(7, 7, 10, NA, 15, NA))
chromPeaks(xchr) <- pks
## Plot the peaks in different colors
plot(xchr, peakCol = c("#ff000080", "#0000ff80"),
peakBg = c("#ff000020", "#0000ff20"))
## Indicate the peaks as rectangles
plot(xchr, peakCol = c("#ff000060", "#0000ff60"), peakBg = NA,
peakType = "rectangle")
 ## Filter the XChromatogram by retention time
xchr_sub <- filterRt(xchr, rt = c(4, 6))
xchr_sub
#> Object of class: XChromatogram
#> length of object: 3
#> from file:
#> mz range: [NA, NA]
#> rt range: [4, 6]
#> MS level: 1
#> Identified chromatographic peaks (1):
#> rt rtmin rtmax into maxo sn
#> 4 2 9 93 19 NA
plot(xchr_sub)
## Filter the XChromatogram by retention time
xchr_sub <- filterRt(xchr, rt = c(4, 6))
xchr_sub
#> Object of class: XChromatogram
#> length of object: 3
#> from file:
#> mz range: [NA, NA]
#> rt range: [4, 6]
#> MS level: 1
#> Identified chromatographic peaks (1):
#> rt rtmin rtmax into maxo sn
#> 4 2 9 93 19 NA
plot(xchr_sub)
